
Abstract
Summary: Spontaneous CSF otorrhea is a relatively rare entity. Adequate diagnosis and treatment are needed to avoid life-threatening complications such as meningitis. Because diagnosis is based on CT findings, identification of the different pathways of congenital fistulas requires detailed knowledge of embryology and anatomy. The facial canal, petromastoid canal, and tympanomeningeal (Hyrtl’s) fissure can be responsible for CSF fluid otorrhea in the absence of any abnormality of the adjacent membranous labyrinth. We report the first documented and imaging case of Hyrtl’s fissure and its treatment.
CSF otorrhea is a rare and potentially dangerous condition, especially because of the risk of meningitis. The most common causes are traumatic injuries (even due to cerebellopontine angle surgery) and chronic ear disease (infectious or neoplastic disease). These acquired forms are far more common than congenital forms.
Congenital CSF otorrhea results from a fistulous connection between subarachnoid space and the tympanomastoid cavity. They may occur through an abnormal labyrinth such as Mondini dysplasia or through a pathway distant from a normal labyrinth. Causes of fistulas associated to a normal labyrinth are the tegmen tympani defects, the petromastoid canal, a wide cochlear aqueduct, the facial canal, and the tympanomeningeal (Hyrtl’s) fissure. Hyrtl’s fissure is congenital and infralabyrinthic cleft that has been incriminated to be responsible for CSF fistulas (1), although our search of the literature would indicate this has never been documented. To our knowledge, only one clinical case of tympanomeningeal fissure has been reported (2). We report the first documented case of a CSF leak by abnormal persistence of tympanomeningeal fissure.
Case Report
A 6-year-old girl was admitted to our otorhinolaryngology department in August 1999 because of clear fluid otorrhea from her right ear. B-type brachydactyly (Bell’s classification) had been diagnosed at birth, after an uncomplicated full-term pregnancy, and genealogical investigation confirmed this dominant autosomal disease in the family. At 2 years old, the patient underwent ureteronephrectomy for right pyelic duplication.
Otorrhea occurred after bilateral myringotomy for treatment of serous otitis media at 20 months old. Clear fluid drained from the right myringotomy for 2 days, and CT findings of the brain and temporal bones were interpreted as normal.
In our department, otoscopic examination showed clear effusion and a lower implantation of the left auricle. Tympanometry indicated C type, and audiometry detected 30-decibal conductive hearing loss in the right ear.
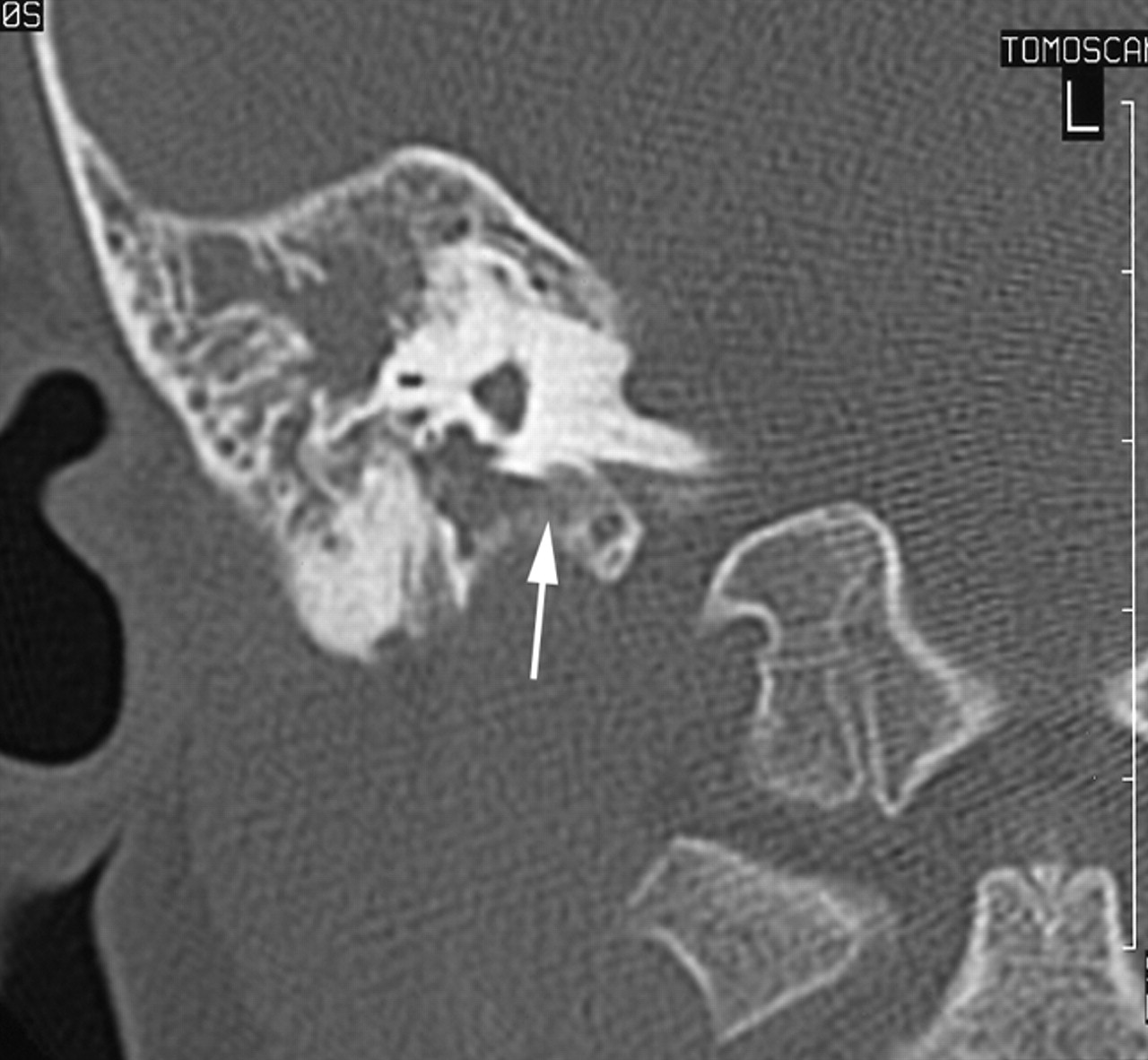
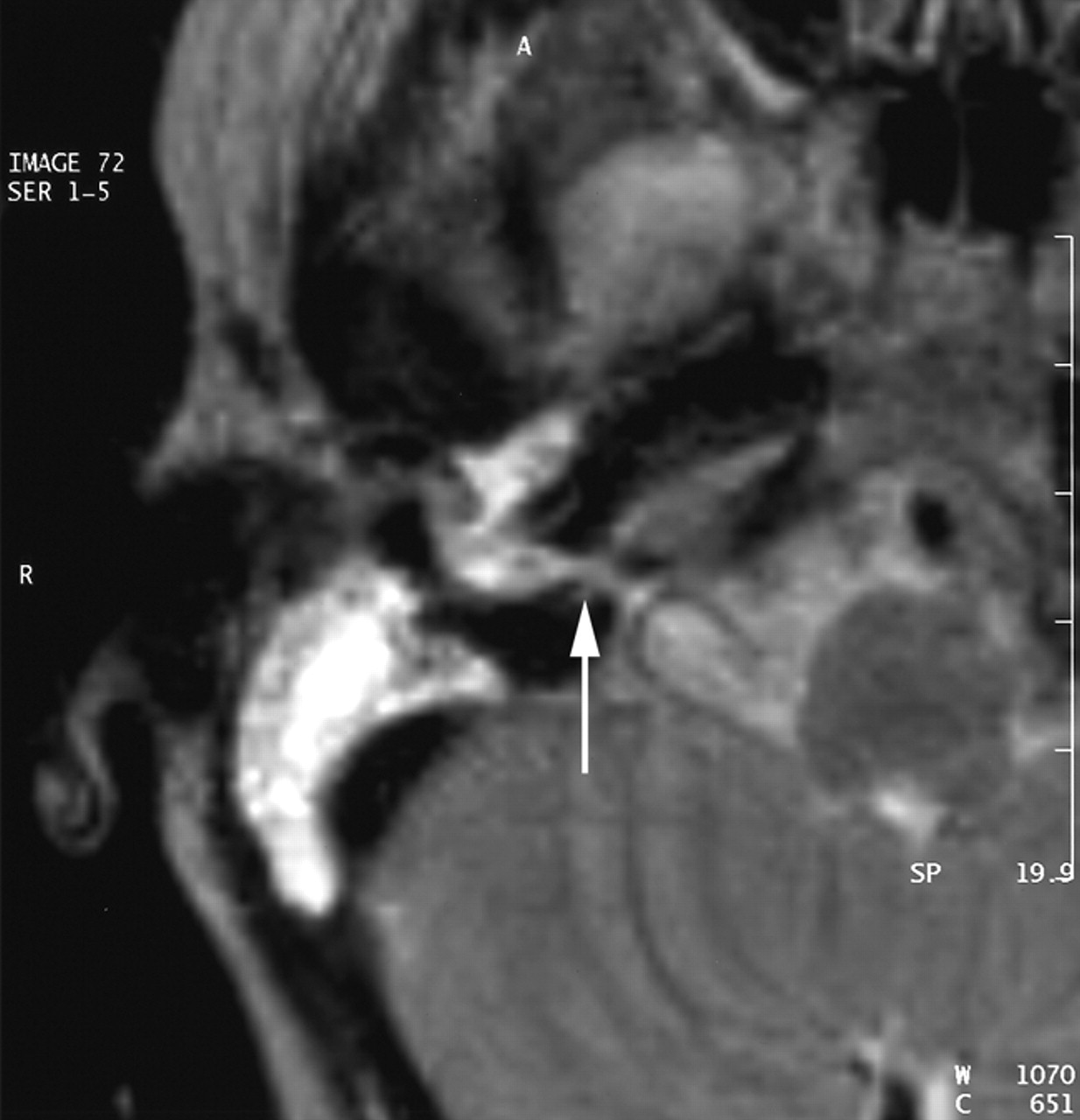
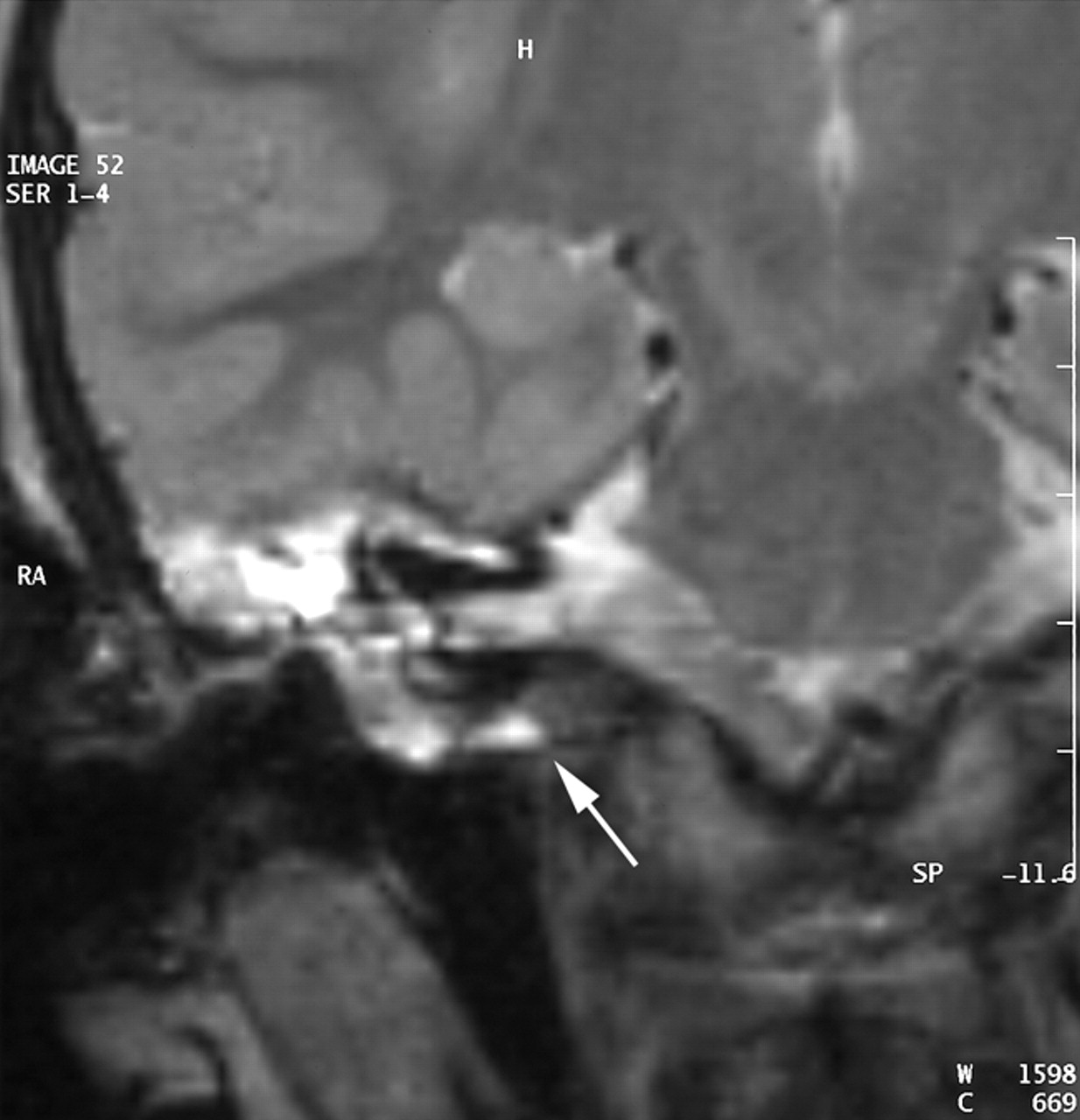
A CT scan of temporal bones revealed a large communication between the subarachnoid space of the cerebellopontine angle and the right hypotympanum anterior to the jugular fossa and below the cochlear aqueduct, from which it appeared to be distinct. The cleft extended between the bony labyrinth and the jugular bulb and toward the posterior fossa close to the endolymphatic sac. Petrous and mastoid cells were opacified. No abnormalities of the tegmen, facial canal, stapes, or vestibule were observed, and the left temporal bone was normal (Figs 1 and 2). MR imaging confirmed these findings and showed no abnormalities of the membranous labyrinth or cochlear aqueduct structure (Figs 3 and 4).

CT scan in the axial plane. The cleft extends between the bony labyrinth and the jugular bulb.

CT scan in the coronal plane. The cleft extends from the hypotympanum, inferior to a normal bony labyrinth, and toward the posterior fossa close to the endolymphatic sac.

MR image in the axial plane obtained with a T2-weighted sequence (TE, 128 TR, 4416; NEX, 1; section thickness, 3 mm). Hyperintensity of the CSF can be seen in the medial cavity communicating with cerebellopontine angle.

MR image in the coronal plane obtained with a T2-weighted sequence (TE, 128; TR, 4416; NEX, 1; section thickness, 3 mm). The cleft is seen under the cochlea and below the internal auditory meatus level.
A surgical approach was performed under general anesthesia. A large endaural incision was made, and a herniation containing CSF was found in the middle ear. The uncudostapedial joint was separated. After incision of the herniation, the aspiration of the fluid contents revealed a communication below the round window. Some fragments of the herniation were removed for histologic analysis. Temporalis muscle fascia was used to fill the cleft and stop CSF leakage. Bone filling was completed by using a mixture of hydroxyapatite granules and fibrin glue. Silastic was then added to maintain the compactness of the mixture.
The patient had an uneventful postoperative course, without further CSF leakage. The only residual symptom was right conductive hearing loss. Histologic analysis showed no meningeal tissue in the middle ear herniation, but only respiratory mucosa. Ossiculoplasty was performed 3 months later to restore tympano-ossicular function. The Silastic was removed, and no leakage was observed.
At 10 years old, the patient is asymptomatic, without leakage, and with normal hearing.
Discussion
To our knowledge, this case is the first documented and imaging case of Hyrtl’s fissure. Hyrtl’s fissure is a possible route for perilabyrinthine congenital fistula. In the 1890s, Siebemann, cited by Spector et al (1), described the gross anatomy of the three structures of temporal bone embryogenesis: the cochlear aqueduct, the first accessory canal containing the inferior cochlear vein (canal of Cotugno), and the second accessory canal described by Hyrtl in 1936 (3). Anton and Bast renamed Hyrtl’s fissure “the tympanomeningeal fissure or hiatus” (1). Recent research by Spector et al (1) provided precise data on the later developmental stages of the periotic duct, as well as indirect evidence for the patency of Hyrtl’s fissure. In the 16–18-week-old fetus, the fossula of the round window is continuous with the posterior cranial fossa, with a direct and functional communication—the tympanomeningeal or Hyrtl’s fissure (1, 4). Between 24 and 26 weeks in utero, the cartilaginous bar is entirely replaced by bone, closing the tympanomeningeal fissure. For Spector et al, this ossification is occasionally incomplete, and permanent tympanomeningeal fissure persists. Very few cases of such persistence have been described in adults, and the case reported in the present study is only the second in a child. Because this malformation is not always functional, its frequency may be underestimated. In 1979, Gacek and Leipzig described a previously unreported case of meningocele through Hyrtl’s fissure, which was revealed by myringotomy and diagnosed by hypocycloidal tomography (2). Another radiological case of Hyrtl’s fissure was reported by Gacek et al in 1999 (5).
Spontaneous cerebrospinal fistula is a difficult diagnosis sometimes revealed only by recurrent attacks of meningitis that may occur in 92% of such fistulas (6), which indicates the importance of diagnosis and treatment of CSF leakage. Recurrent meningitis, clear otorrhea, or rhinorrhea are signs requiring several investigations of the temporal bone. In our case, no meningitis occurred. When the ear drum is intact, CSF passes down the eustachian tube and may result in rhinorrhea. If the tympanic membrane is perforated, either spontaneously or after myringotomy, otorrhea may occur (7). As in our case, some reported cases have already shown that congenital CSF leakage may present as serous otitis media and be revealed at the time of myringotomy (4). The beta-2-transferrin band is a reliable method for diagnosing CSF otorrhea (8). CT involving 1-mm sections in coronal and axial planes of the temporal bone is certainly the most precise and reliable method available. The first CT scan of our patient showed no abnormalities, probably because of the poor parameters used (e.g., planes too spaced out and unawareness of the different preformed bone pathways that can be incriminated in such a disease). A second CT scan showed an enlarged communication between the posterior fossa near the posteroinferior cerebellar artery and the hypotympanum posteroinferior to the round window. The fistula was seen to be lower and distinct from the cochlear aqueduct, which appeared to be normal (Figs 1 and 2). MR imaging showed that the cochlea structure and the membranous labyrinth were normal and confirmed the suspected presence of CSF in the right middle ear. Adjacent temporal bone was free of abnormality (Figs 3 and 4).
Spontaneous congenital CSF fistulas can be divided into two groups relative to age and pathogenicity (8). For the first type, the congenital defect becomes apparent in adulthood. Twenty-eight percent of the spontaneous CSF reported cases are the adult type. It is characterized by multiple (8–15) bone defects 1–6 mm in diameter, most commonly of the tegmen tympani or tegmen mastoideum. The possible pathogenic mechanisms included progressive sagging and rupture of the dura due to congenital tegmen dehiscence and progressive bone erosion by aberrant arachnoid granulations (7). According to Gacek et al, these granulations form at birth and may enlarge with age and erode the bone margins of pneumatized parts of the skull (5, 9).
The second type is usually diagnosed early in life, between ages 1 and 5 years. CSF communicates with middle ear spaces through a preformed bone pathway (5, 6). This pathway can pass through the labyrinthine window as a result of erosion or around the labyrinth because of dural herniation in the middle ear (4, 5). Neely suggested a classification into three types (10): type I, abnormal connections through the otic capsule; type II, abnormal connections adjacent to the otic capsule; and type III, abnormal connections distant from the otic capsule. The diagnosis and therapeutic implications of each type are unique.
The most common route for congenital CSF otorrhea is translabyrinthine (11). The communication passes from the internal auditory meatus through the modiolus and then into a dysplastic labyrinth where stapes deformity frequently allows fluid leakage. The stapes footplate is commonly malformed because of either a common embryologic defect or erosion from continuous pulsatile CSF pressure (12). The most commonly reported malformation is Mondini dysplasia. Other labyrinthine malformations have been clearly described (11). Congenital CSF leakage through a well-developed labyrinth has been reported in which case the route is an enlarged cochlear aqueduct (6, 12). Several reports of supposed cochlear aqueduct enlargement showed images of a dilated medial aperture well within the range of normal variability (13). Many studies, however, have demonstrated the passage of CSF into the perilymph, which would indicate that the cochlear aqueduct is patent (1).
Extralabyrinthine but adjacent (type II of Neely) route for congenital CSF leakage is less common. A probable prerequisite for fistula adjacent to the labyrinth is abnormal enlargement of a preformed bone pathway (4). The three possible sites for fistula around the labyrinth are the petromastoid canal, the facial canal, and Hyrtl’s fissure. The petromastoid canal, which contains the subarcuate artery and vein (14), begins in the subarcuate fossa above and lateral to the internal auditory canal, passes under the arch of the superior semicircular canal and then above the horizontal semicircular canal, before terminating its course in the cells surrounding the mastoid antrum. Congenital leakage through this canal has been reported elsewhere (4). The initial segment of the facial canal has also been incriminated as a pathway for congenital CSF leakage, and four cases have been reported (4, 15–18).
Unlike acquired fistulas, which can resolve spontaneously, spontaneous congenital CSF leakage requires surgical treatment. Various drainage, packing, and obliteration procedures with middle cranial fossa or transmastoid approaches have been developed in an attempt to stop CSF leakage and prevent meningitis. Some authors have suggested that osteoconductive biomaterials are better than traditional methods (19). With such bone substitutes, craniotomy is unnecessary. In our patient, fistula could be filled with a composite mixture of fibrin glue and biphasic calcium phosphate ceramic. An uneventful 4-year postoperative period attests to the validity of this surgical procedure.
Conclusion
Because this disorder is rare, misdiagnosis or failure to make a timely early diagnosis is common, which means that suitable therapy may be delayed. Better knowledge of the possible sites and pathways of fistulas (even rare ones) is necessary. The different pathways of spontaneous CSF leakage should be clearly understood and carefully examined by the radiologist and the surgeon. Hyrtl’s fissure is a possible, although very infrequent, fistula route, which can be misdiagnosed even when CT is performed. The treatment for this congenital fistula is based on filling of the bone pathway, which can be done with biomaterials.
References
- Received March 18, 2004.
- Accepted after revision June 4, 2004.
- American Society of Neuroradiology


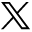


{kind=link}
{kind=link}
{kind=link}
{kind=link}