
Abstract
SUMMARY: Polymorphous low-grade neuroepithelial tumors of the young (PLNTY) are rare brain tumors first described in 2017 and recently included in the 2021 5th World Health Organization Classification of Tumors of the Central Nervous System. They typically affect children and young adults. Few pediatric cases have been reported in the literature. The most common imaging features described, include location within the temporal lobe, involvement of the cortical/subcortical region, coarse calcifications, and well-defined margins with solid and cystic morphology, with slight-or-no enhancement. However, there is limited information on imaging features in children. We present the imaging spectrum of neuroimaging features in a series of pediatric patients with a histologically and molecularly proved PLNTY diagnosis. Coarse calcifications are uncommon in children compared with the adult literature, and they may develop with time. The transmantle-like sign can be observed, and adjacent cortical dysplasia may be seen. Seizure recurrence may occur despite gross total resection of the tumor.
ABBREVIATIONS:
- DNET
- dysembryoplastic neuroepithelial tumor
- IQR
- interquartile range
- PLNTY
- polymorphous low-grade neuroepithelial tumor of the young
- TLS
- transmantle-like sign
- WHO5
- World Health Organization Classification of Tumors of the Central Nervous System, 5th edition
Polymorphous low-grade neuroepithelial tumor of the young (PLNTY) was described in 2017 by Huse et al1 as an epileptogenic tumor with infiltrative growth, oligodendroglioma-like components, frequent calcification, strong diffuse aberrant CD34 expression, and genetic alterations in the mitogen-activated kinase pathway, including FGFR2, FGFR3, and BRAF.1⇓-3
PLNTY is 1 of the 6 newly recognized pediatric low‐grade gliomas and glioneuronal tumors in the 2021 5th edition of the World Health Organization Classification of Tumors of the Central Nervous System (WHO5).4 It primarily affects children and young adults2 but can also be seen in individuals older than 30 years of age.5,6
Few cases have been reported, including adult and pediatric cases.7,8 The imaging features of this entity have been shown to be diverse,3,9 and calcifications are recognized as an important imaging characteristic of PLNTY.8,10 However, there is still scarce information on the imaging features in children.
PLNTY has a benign clinical course with few exceptions.8,11 Correct identification of PLNTY can affect patient treatment and prognosis by directing surgical management and medical treatment. We aimed to describe the neuroimaging features in a series of pediatric patients with a histologic and molecularly-proved PLNTY diagnosis.
CASE SERIES
This single-center, retrospective study was reviewed and approved by our institutional review board and complied with the Health Insurance Portability and Accountability Act. We obtained a waiver of informed consent. We searched our databases (mPower by Nuance Communications, https://www.nuance.com/healthcare/diagnostics-solutions/radiology-performance-analytics/mpower-clinical-analytics.html;, and Illuminate InSight, https://www.illuminate.ai/illuminate/solutions/insight) for pathology-proved PLNTY cases. We extracted demographic data from an electronic chart system (Epic Systems, https://www.forbes.com/companies/epic-systems/?sh=627bbf05ca11). Inclusion criteria were 0–18 years of age, availability of preoperative brain MR imaging, and a molecular diagnosis based on immunohistochemical or genetic analysis.
We evaluated 10 patients. Five (50%) were female. The median age at diagnosis was 9 years (range, 0–14 years). The most common symptoms at presentation were seizures (n = 8, 80%). The median duration of symptoms was 32 months (interquartile range [IQR], 5.75–85 months). All tumors were surgically resected. The median time interval between MR imaging and surgery was 9.5 months (IQR, 2.75–59.5 months). Recurrence of seizure symptoms was seen in 3 patients (30%), who required surgical reintervention. Demographics, clinical data, and pathologic features of this series are summarized in the Online Supplemental Data.
Imaging
Patients were referred from different health centers, and initial imaging protocols were not fully standardized. The patients had undergone either 1.5T or 3T brain MR imaging (Various scanners; Siemens). Sequences performed included sagittal and axial T1WI, axial T2WI, T2 FLAIR, DWI, and gadolinium-enhanced postcontrast T1WI. Gradient-echo, SWI, and arterial spin-labeling sequences were variably obtained depending on the study and scanner. All imaging from our institution included 0.9- to 1.0-mm isotropic T1-weighted MPRAGE and high-resolution 2-mm (no gap) T2-weighted and 3-mm T2 FLAIR images in the axial and coronal planes.
Two pediatric neuroradiologists systematically evaluated all imaging in consensus to assess the following: tumor location, margins, and morphology; T1WI, T2WI, susceptibility imaging, T2 FLAIR signal; and diffusion characteristics and contrast enhancement. If available, CT attenuation was also evaluated.
All tumors were supratentorial. Most tumors involved the temporal lobe (n = 7, 70%) with a mixed cortical-subcortical location (n = 8, 80%). The most common tumor morphology was a mixed solid and cystic pattern (n = 8, 80%). The median cyst size was 5.5 mm (IQR, 3.2–8.7 mm). The largest measured tumor was 17.2 mm. Most tumors had no (n = 6, 60%) or mild (n = 4, 40%) contrast enhancement. Margins were operationally defined on T2WI as a clear demarcation between the tumor and the surrounding brain tissue for most of the circumference of the tumor margin. Nine (90%) tumors had ill-defined margins. No restricted (reduced) diffusion was seen in the tumors. Calcifications were determined by a combination of T1-weighted, T2-weighted, SWI with unwrapped phase images, gradient recalled-echo, and CT on initial and follow-up imaging. Two (20%) cases had initial CT. Eight tumors (80%) had visible calcifications, predominately with a punctate pattern (n = 5, 50%) (Figure and Table). One case (case 4) showed a gradual development of a large central calcification during 10 years (Online Supplemental Data). A transmantle-like sign (TLS) was defined as a linear T2 or T2 FLAIR hyperintense signal in the white matter from the mass that tapers toward the lateral ventricles. This finding was seen in 4 (40%) patients (Fig 1J, -L). When available, a mixed pattern of low or only mildly elevated perfusion was seen on arterial spin-labeling. Imaging findings are summarized in the Online Supplemental Data.

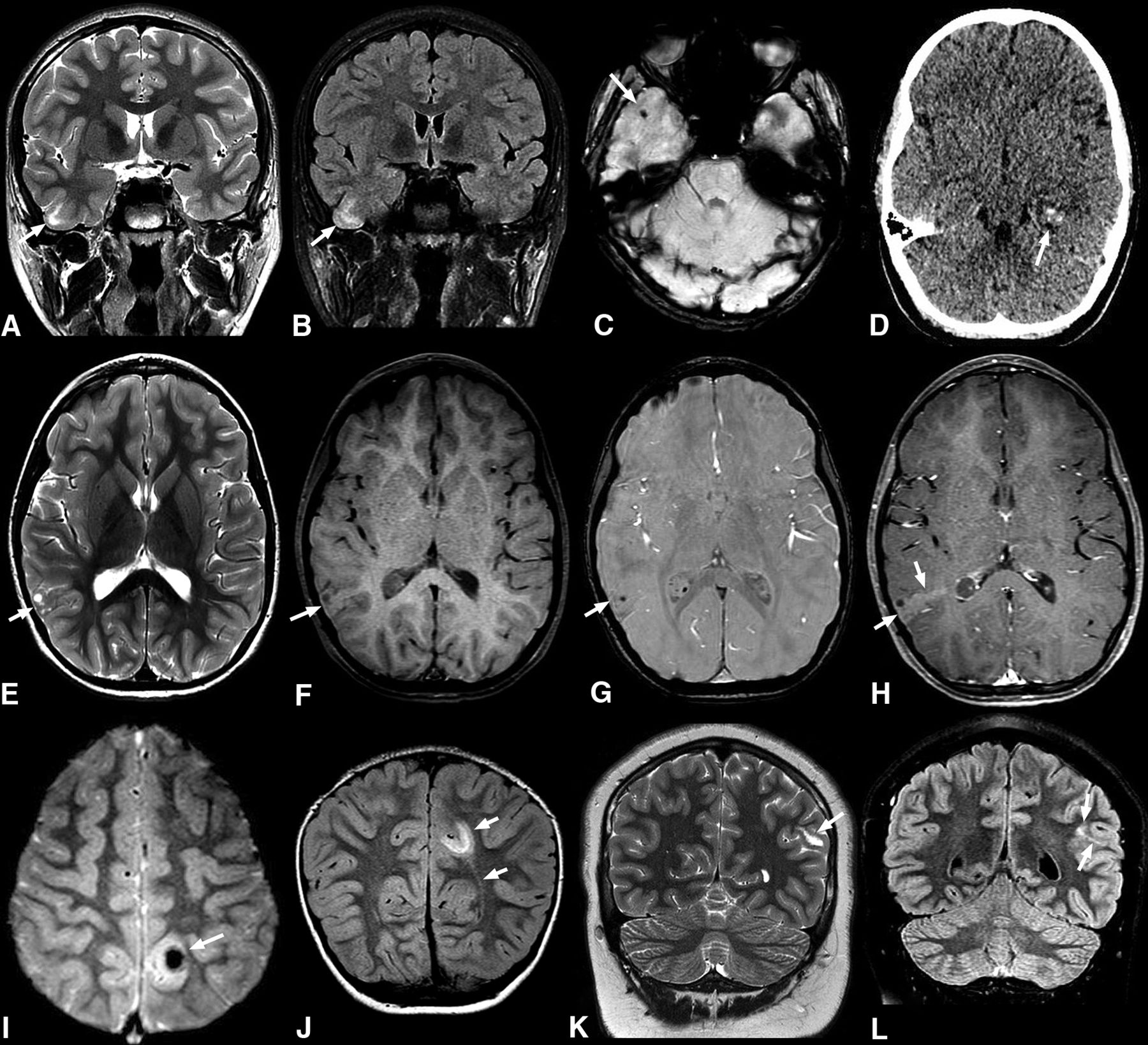
Common imaging features of pediatric PLNTY tumors. A–C, A 9-year-old boy (case 5) with a 1-month history of headaches. Coronal T2WI (A) and FLAIR (B) sequences show a vague area of increased signal intensity in the cortical-subcortical region of the right temporal lobe (arrows in A and B). Axial SWI shows a small calcification (C). D, An 11-year-old girl with seizures (case 1). Nonenhanced axial CT shows a focal lesion in the left temporal lobe with intralesional calcifications exhibiting a punctate calcification pattern (arrow). E–H, A 6-year-old girl with complex focal seizures (case 3). Axial T2WI (E) image depicts a mixed cortical-subcortical lesion with solid and cystic components in the right parietal lobe. Pre- and postcontrast axial T1 TSE (F and H) reveals mild postcontrast enhancement (arrows in E and F). Axial SWI (G) demonstrates a punctate pattern of calcification (arrow). I and J, A 4-year-old girl with a 4-year history of seizures (case 6). Axial T2* gradient-echo image (I) reveals an ill-defined lesion in the left parietal lobe with a large central susceptibility in keeping with a chunky central calcification (arrow). Coronal FLAIR image (J) shows a related high-signal-intensity area in the subcortical white matter extending toward the left lateral ventricle, consistent with a TLS (arrows). K and L, A 14-year-old boy with new-onset seizures (case 8). Coronal T2WI (K) shows a hyperintense lesion in the left parietal region (arrow). Coronal FLAIR (L) shows an associated high signal area extending medially, in keeping with a TLS (arrows).
| Percentage, Number | |
|---|---|
| Location | |
| Temporal lobe | 70%, n = 7 |
| Parietal lobe | 20%, n = 2 |
| Occipital lobe | 10%, n = 1 |
| Cortical-subcortical | 80%, n = 8 |
| Subcortical only | 20%, n = 2 |
| Morphology | |
| Mixed solid and cystic | 80%, n = 8 |
| Solid | 10%, n = 1 |
| Cystic | 10%, n = 1 |
| Calcifications | |
| Punctate | 50%, n = 5 |
| Chunky | 30%, n = 3 |
| No definite Ca2+ on imaging | 20%, n = 2 |
| Margins | |
| Ill-defined | 90%, n = 9 |
| Well-defined | 10%, n = 1 |
| Contrast enhancement | |
| Mild | 40%, n = 4 |
| None | 60%, n = 6 |
| TLS | |
| Yes | 40%, n = 4 |
| None | 60%, n = 6 |
| Pathology proven cortical dysplasia | 30%, n = 3 |
| Pathology proven calcification | 70%, n = 7 |
| Mutation | |
| FGFR2 alteration | 50%, n = 5 |
| BRAF mutation | 50%, n = 5 |
Distribution of imaging and pathologic features
Pathologic Features
Tumors diagnosed as PLNTY were reviewed by a board-certified neuropathologist, blinded to clinical and radiologic data. The evaluation included a review of both hematoxylin & eosin–stained slides and immunohistochemical stains. In addition, next-generation sequencing was performed on all tumors. Tumors that fulfilled the WHO5 “essential criteria” for PLNTY4 were selected for inclusion. The WHO criteria are summarized in the Online Supplemental Data. Two additional “desirable criteria” (calcifications and absence of 1p/19q codeletion)4 and the presence of adjacent cortical dysplasia on histology were also assessed.
All tumors met all the WHO5 essential criteria as well as the desirable criterion of lacking 1p/19 codeletion. Seven cases (70%) showed calcifications on histology. All tumors showed at least focal diffuse growth and oligodendroglial-like components. All tumors showed areas of positive staining for glial fibrillary acidic protein and/or the oligodendrocyte transcription factor. Intense, diffuse CD34 staining was present in all cases, as well as regions of ramified staining; and the Ki-67 proliferation indices ranged from <1% to 3%–5%. Mitotic activity was typically absent, though 1 tumor demonstrated 3 mitoses per 10 high-power fields. Necrosis and microvascular proliferation were not seen in any case. Calcifications ranged from diffuse to sparse or absent. Histologic features of cortical dysplasia were present in 3 cases (30%). We had 5 cases of FGFR2 fusions and 5 cases of BRAF p.V600E mutations. Pathologic findings are shown in the Online Supplemental Data.
DISCUSSION
To our knowledge, this is the largest series of imaging features of the rare tumor entity PLNTY in children. In our study, all tumors were in the supratentorial compartment, and most were in the temporal lobe in a cortical-subcortical location, in keeping with prior literature.5,7,9 Kurokawa et al,9 in a systematic review, reported 2 main imaging patterns for PLNTY: 1) a well-circumscribed, solid, and cystic mass with scarce mass effect surrounded by normal parenchyma; and 2) an ill-defined, small mass with tiny cysts and TLS. Although the first pattern was reported as the most common in that article, in this study, the second pattern was predominant.
Prominent coarse and central calcifications have been suggested as characteristic features of PLNTY.3 However, extensive calcifications were not a common imaging feature in this series. Calcifications showed, predominately, a punctate pattern, also called “salt and pepper pattern.”12 Despite these differences, 1 case gradually developed a sizeable central calcification during a follow-up period of 10 years, exhibiting the classic prominent central calcification. This finding suggests that there may be an evolving imaging appearance of these tumors, in which small calcifications develop in younger patients and grow or coalesce with time to become dense coarse calcifications in older patients. Due to the small number of cases, we cannot assess a correlation between the age of presentation or the duration of symptoms with the presence or development of prominent central calcifications. For example, our case 6 with a 4-year history of seizures had a prominent calcification on the initial MR imaging, while our case 10 did not demonstrate calcifications on the initial MR imaging or develop calcifications during 7 years.
Kurokawa et al9 associated TLS with a longer duration of symptoms and found that cortical dysplasia was more frequent in patients with TLS. Other reports have also described this association.10,13 In our study, the duration of seizures for cases that showed TLS was variable, and pathology-proved cortical dysplasia in association with TLS was found in only 25% (1 of 4) of patients. These findings may be due to pathologic sampling confounding. It is also possible that cortical dysplasia may be seen without a clear TLS, or alternatively, not all TLSs are truly indicative of cortical dysplasia. The proximity of temporal lobe tumors to the margins of the ventricles may also cause difficulties in appreciating the TLS. A transmantle appearance may cause diagnostic confusion on imaging, with the possibility of assigning simple focal cortical dysplasia type IIb as the diagnosis, or in cases in which a mass is readily apparent, the tumor may be diagnosed as dysembryoplastic neuroepithelial tumor (DNET). However, the features of PLNTY as described here can indicate a correct diagnosis or a more targeted differential consideration of PLNTY in children. Close attention to the presence of small tumoral features on multiple planes can prevent this potential misdiagnosis.
Isolated reports describe PLNTY cases with difficult therapeutic control or disease recurrence.11,14 Armocida et al8 found that contrast enhancement in PLNTY was associated with recurrence or poor control of epileptic symptoms. We found a recurrence of seizures after surgery in 3 cases; none showed postcontrast enhancement. We suspect that the presence of contrast enhancement is not necessarily the main predictor of poor seizure control.
Nearly all the PLNTYs have either BRAF p.V600E mutations or fusion events involving FGFR2/FGFR3.1 The BRAF p.V600E mutation is the most common.5 We had an equal proportion of BRAF p.V600E mutations and FGFR2 fusions. There are no clear associations between genetic mutations and clinical outcomes in patients with PLNTY.8 We found that recurrence of symptoms after surgery occurred similarly in patients with both mutations. The FGFR3-TACC3 fusion gene in PLNTY has shown malignant transformation in some reports.15 This mutation was not found in our series.
The differential diagnosis of PLNTY in children includes DNET, ganglioglioma, and pleomorphic xanthoastrocytoma, all of which are commonly seen in the temporal lobe and can commonly present with seizures.6,9,10 Cystic changes and calcifications have been reported to be more common in PLNTY than in gangliogliomas,16 though we do not find that this distinction helped guide a differential diagnosis. Nevertheless, larger unilocular cysts are not typical of PLNTY and were not seen in our series. Vasogenic edema is uncommon in all these tumors, and they may be seen in cortical/subcortical locations. PLNTY may show mild or no contrast enhancement, differing from the typical moderate-to-marked enhancement in parts of ganglioglioma or pleomorphic xanthoastrocytoma. Intratumoral T2 FLAIR hyperintense septations with multiple cystlike central T2 FLAIR hyperintense areas (FLAIR rim sign) are much more frequent in DNET.9 Both DNET and ganglioglioma may have associated cortical dysplasia on histopathology. Neurocysticercosis also has cystic areas with punctate calcifications but typically does not have a clear associated mass.
CONCLUSIONS
Unlike previous reports, which have mostly been documented in adult cases, large, dense calcifications and well-defined margins are not commonly seen in pediatric cases. We also suggest that these tumors may have a slowly evolving imaging appearance. PLNTY can show associated cortical dysplasia on pathology, not always associated with a TLS. Seizure recurrence may occur despite gross total resection.
Footnotes
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
References
- Received October 17, 2023.
- Accepted after revision December 16, 2023.
- © 2024 by American Journal of Neuroradiology




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.