
Abstract
BACKGROUND AND PURPOSE: Neuronal ceroid lipofuscinoses are a group of neurodegenerative disorders characterized by the accumulation of autofluorescent lipopigments in neuronal cells. As a result of storage material in the brain and retina, clinical manifestations include speech delay, cognitive dysfunction, motor regression, epilepsy, vision loss, and early death. At present, 14 different ceroid lipofuscinosis (CLN) genes are known. Recently, the FDA approved the use of recombinant human proenzyme of tripeptidyl-peptidase 1 for CLN2 disease, while phase I/IIa clinical trials for gene therapy in CLN3 and CLN6 are ongoing. Early diagnosis is, therefore, key to initiating treatment and arresting disease progression. Neuroimaging features of CLN1, CLN2, CLN3, and CLN5 diseases are well-described, with sparse literature on other subtypes. We aimed to investigate and expand the MR imaging features of genetically proved neuronal ceroid lipofuscinoses subtypes at our institution and also to report the time interval between the age of disease onset and the diagnosis of neuronal ceroid lipofuscinoses.
MATERIALS AND METHODS: We investigated and analyzed the age of disease onset and neuroimaging findings (signal intensity in periventricular, deep, and subcortical white matter, thalami, basal ganglia, posterior limb of the internal capsule, insular/subinsular regions, and ventral pons; and the presence or absence of supratentorial and/or infratentorial atrophy) of patients with genetically proved neuronal ceroid lipofuscinoses at our institution. This group consisted of 24 patients who underwent 40 brain MR imaging investigations between 1993 and 2019, with a male preponderance (male/female ratio = 15:9).
RESULTS: The mean ages of disease onset, first brain MR imaging, and diagnosis of neuronal ceroid lipofuscinoses were 4.70 ± 3.48 years, 6.76 ± 4.49 years, and 7.27 ± 4.78 years, respectively. Findings on initial brain MR imaging included T2/FLAIR hypointensity in the thalami (n = 22); T2/FLAIR hyperintensity in the periventricular and deep white matter (n = 22), posterior limb of the internal capsule (n = 22), ventral pons (n = 19), and insular/subinsular region (n = 18); supratentorial (n = 21) and infratentorial atrophy (n = 20). Eight of 9 patients who had follow-up neuroimaging showed progressive changes.
CONCLUSIONS: We identified reported classic neuroimaging features in all except 1 patient with neuronal ceroid lipofuscinoses in our study. CLN2, CLN5, and CLN7 diseases showed predominant cerebellar-over-cerebral atrophy. We demonstrate that abnormal signal intensity in the deep white matter, posterior limb of the internal capsule, and ventral pons is more common than previously reported in the literature. We report abnormal signal intensity in the insular/subinsular region for the first time. The difference in the median time from disease onset and diagnosis was 1.5 years.
ABBREVIATIONS:
- CLN
- ceroid lipofuscinosis
- DWM
- deep white matter
- IQR
- interquartile range
- I-SI
- insular/subinsular region
- NCL
- neuronal ceroid lipofuscinoses
- PLIC
- posterior limb of the internal capsule
- PVWM
- periventricular white matter
- SCWM
- subcortical white matter
Neuronal ceroid lipofuscinoses (NCL) are a group of clinically and genetically heterogeneous lysosomal storage disorders characterized by the accumulation of autofluorescent lipopigments in neuronal cells of the brain and retina.1 They constitute the most common neurodegenerative disorders of childhood, with an estimated incidence of 1.6–2.4/100,000 in the United States, 2.2/100,000 in Sweden, 2–2.5/100,000 in Denmark, 3.9/100,000 in Norway, 4.8/100,000 in Finland, and 7/100,000 in Iceland.1,2 Clinical manifestations include speech delay, cognitive dysfunction, developmental regression, epilepsy, vision loss, and early death.3 Although traditionally classified on the basis of the age of onset (infantile, late-infantile, juvenile, and adult), classification is now based on genetic etiology, with 14 different ceroid lipofuscinosis (CLN) genes known at present (Table 1).4 Several new insights into the cell biology of NCL have emerged,5⇓-7 with ongoing translational research likely to have therapeutic implications.8 Furthermore, it has been proposed that the different subtypes of NCL are actually distinct disorders, each with a unique underlying molecular pathomechanism sharing a common end point in the form of neuronal loss and autofluorescent storage material.8 Recently, the FDA approved the use of cerliponase alfa (Brineura), a recombinant human proenzyme of tripeptidyl-peptidase 1, for CLN2 disease.9 The treatment arrests disease progression; therefore, early diagnosis is essential for optimal treatment outcomes. In addition, phase I/IIa clinical trials with gene therapy are currently in progress for CLN310 and CLN6 diseases.11
Subgroups of NCL, their genes, protein names, and MIM numbers
The classic neuroimaging features in brain MR imaging of NCL reported in the literature include T2-hypointense thalami, T2-hyperintense periventricular white matter, and progressive cerebral and cerebellar volume loss.12⇓⇓⇓⇓⇓⇓-19 Subtype-specific imaging findings are well-described in CLN1, CLN2, CLN3, and CLN5 diseases,20⇓⇓⇓⇓-25 with scarce literature on other subtypes.26⇓⇓⇓⇓-31 Given the rapidly changing landscape in cell biology and translation in NCL and the need for early diagnosis facilitating early treatment, we performed a retrospective review study to investigate and expand the MR imaging features of the disease in patients with genetically confirmed NCL diagnosed at our institution. We also report the time interval between the age of disease onset, first brain MR imaging, and diagnosis in this study.
MATERIALS AND METHODS
The institutional Research Ethics Board approved the study (REB 1000065260). Patients were identified from a previous study data base.32 Patient demographics and clinical features were obtained through electronic chart review. Brain MR imaging examinations were reviewed for quality and co-read by 2 of the authors (A.B. and P.K.) with consensus review. Specifically, the nature of T2 signal intensity in the thalami, periventricular white matter (PVWM), deep white matter (DWM), subcortical white matter (SCWM), basal ganglia, posterior limb of the internal capsule (PLIC), insular/subinsular region (I-SI), and ventral pons, along with the presence or absence of supratentorial and infratentorial atrophy, was documented. Atrophy was graded subjectively as mild, moderate, and severe. Note was made of the degree of cerebral atrophy relative to cerebellar atrophy. Volumetric analysis was not performed due to insufficient numbers of cases with volume data. The age of disease onset was determined by documented history in the hospital records system (Chartmaxx; https://chartmaxx.software.informer.com/). A working diagnosis of NCL was defined either by a histopathologic basis with positive conjunctival or skin biopsy demonstrating curvilinear profiles, fingerprint profiles, or granular osmiophilic profiles; or by a clinicoradiologic basis when clinical presentation or family history, or both in conjunction with MR imaging findings were highly suggestive of the diagnosis. All patients in the cohort eventually had confirmed genetic diagnoses via direct Sanger testing, the details of which have been published previously.32 Data were entered into Excel (Microsoft). Data were analyzed with descriptive statistics (median and interquartile range [IQR] for age calculated in Excel).
RESULTS
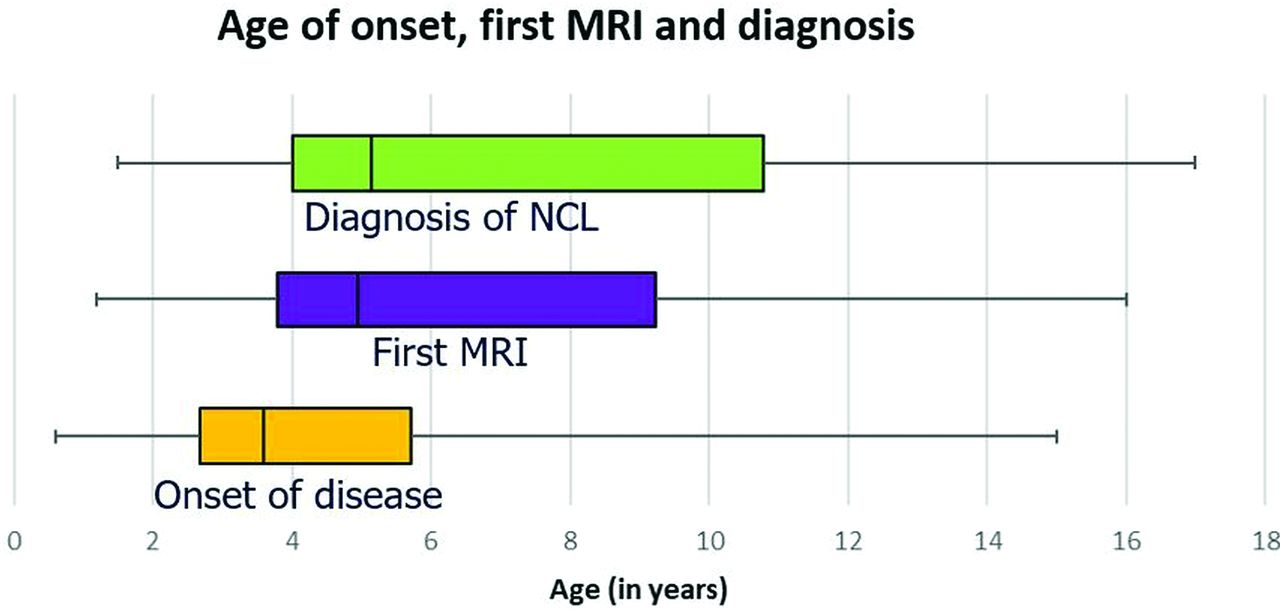
Thirty-four patients with genetically confirmed neuronal ceroid lipofuscinosis were identified between 1993 and 2019. Most of these patients and their molecular genetic investigations have been reported.32 Of these, 24 patients (male/female ratio = 15:9) had 40 MR imaging examinations and were therefore included in the study. The time delay between disease onset and diagnoses is depicted in Fig 1. The median (IQR) age of disease onset, first brain MR imaging, and age of genetic diagnoses were 3.6 (3.0), 4.9 (5.4), and 5.1 (6.7) years, respectively (Fig 1). The NCL subtypes were CLN1 (n = 6), CLN2 (n = 5), CLN3 (n = 2), CLN5 (n = 1), CLN6 (n = 2), CLN7 (n = 6), and CLN8 (n = 2) diseases. The most common symptom was developmental and cognitive regression (n = 22). Other presenting symptoms included speech delay (n = 21), seizures (n = 19), impaired vision (n = 14), and behavioral abnormalities (n = 11). Clinical features and their distribution to each subgroup of NCL are listed in Table 2.

Time delay between disease onset (median, 3.6 years; IQR, 3 years) and clinical, radiologic, and histopathologic diagnoses (median, 5.1 years; IQR, 6.7 years) of NCL is depicted.
Clinical features of patients with NCLa
Brain MR imaging features are summarized in Table 3. Findings on initial brain MR imaging included the following: 1) T2/FLAIR hypointensity in the thalami (n = 22); 2) T2/FLAIR hyperintensity in the PVWM and DWM (n = 22); 3) T2/FLAIR hyperintensity in the PLIC (n = 22); 4) T2/FLAIR hyperintensity in the ventral pons (n = 19); 5) T2/FLAIR hyperintensity in the I-SI region (n = 18); 6) supratentorial atrophy (n = 21; mild, [n = 10]; moderate-severe, [n = 11]; and 7) cerebellar atrophy [n = 20]). Eight of 9 patients who had follow-up neuroimaging showed progressive changes. Imaging findings in each subtype are summarized below.
Brain MR imaging features of patients with NCL
CLN1 Disease
In CLN1 disease (n = 6), the median (IQR) age of onset was 3 (8.5) years, while the median (IQR) age at first MR imaging was 3.5 (11.5) years. On the initial MR imaging, all patients (n = 6) showed T2/FLAIR hypointensity in the thalami and T2 hyperintensity in the basal ganglia. All patients had T2/FLAIR hyperintensity in the PVWM, DWM, PLIC, and pons, while only 1 patient showed abnormality in the SCWM. Four patients demonstrated T2/FLAIR hyperintensity in the I-SI region. All patients showed supratentorial atrophy (mild = 2; moderate = 2; severe = 2) and cerebellar atrophy (mild = 4; moderate = 2), with 5 patients showing brain stem atrophy. In all 6 patients, the degree of cerebral atrophy was greater than that of cerebellar atrophy.
Three of 6 patients had follow-up imaging. All 3 patients demonstrated progressive supratentorial and cerebellar volume loss, and 2 patients showed progressive white matter T2/FLAIR hyperintensity now extending to involve the SCWM.
CLN2 Disease
In CLN2 disease (n = 5), the median (IQR) age of onset was 3.5 (5.3) years. The median (IQR) age the first MR imaging was 8.7 (8.4) years. All patients (n = 5) showed T2/FLAIR hypointensity in the thalami, whereas 3 patients showed T2 hyperintensity in basal ganglia. All patients showed T2/FLAIR hyperintensity in the PLIC, PVWM, and DWM, with 1 patient demonstrating abnormality involving the SCWM. Four patients showed T2/FLAIR hyperintensity in the I-SI region, and 3 showed signal abnormality in the pons.
Four patients showed supratentorial atrophy (mild = 1; moderate = 1; severe = 2) and cerebellar atrophy (mild = 2; severe = 2), with 4 patients showing brain stem atrophy. In 3 of 5 patients, cerebellar atrophy was greater than cerebral atrophy.
Three patients had follow-up imaging. All 3 showed progressive supratentorial atrophy, and 2 showed progression in cerebellar atrophy. One patient who had normal signal in the I-SI region and pons on the initial MR imaging showed signal abnormality in both regions on follow-up MR imaging.
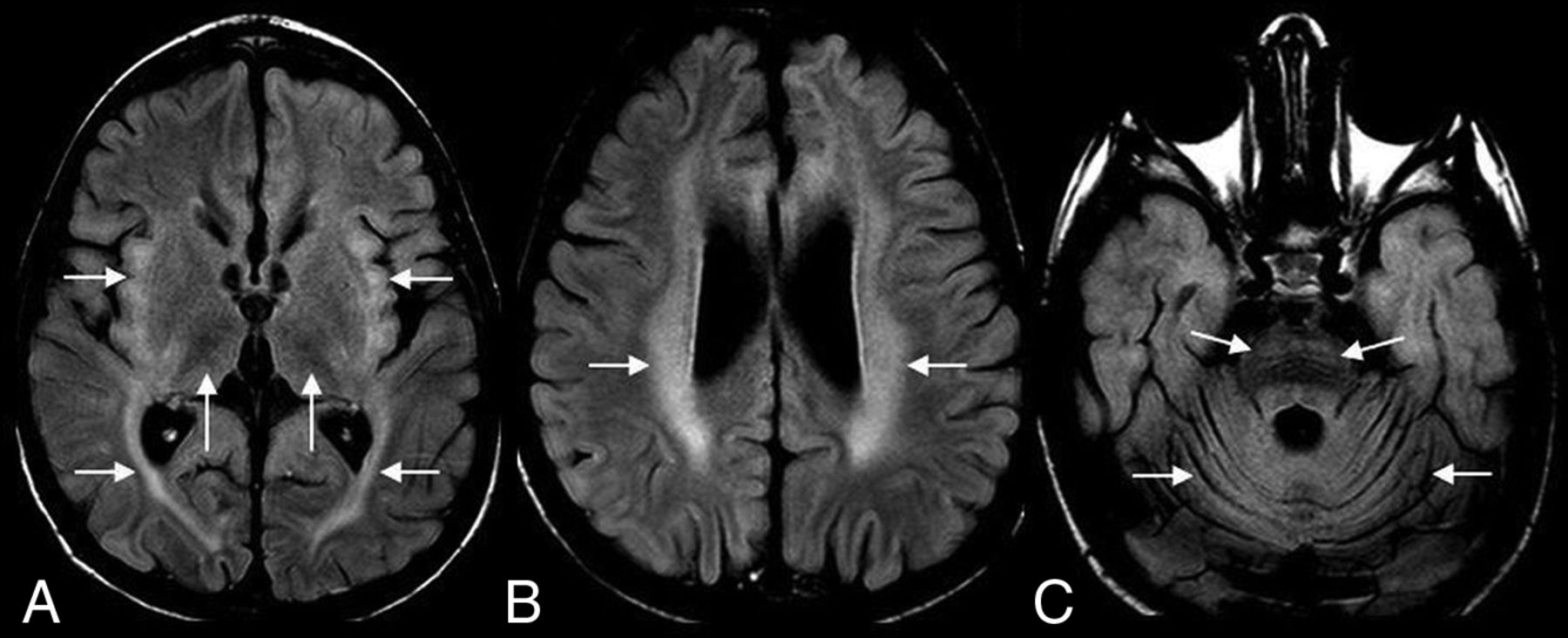
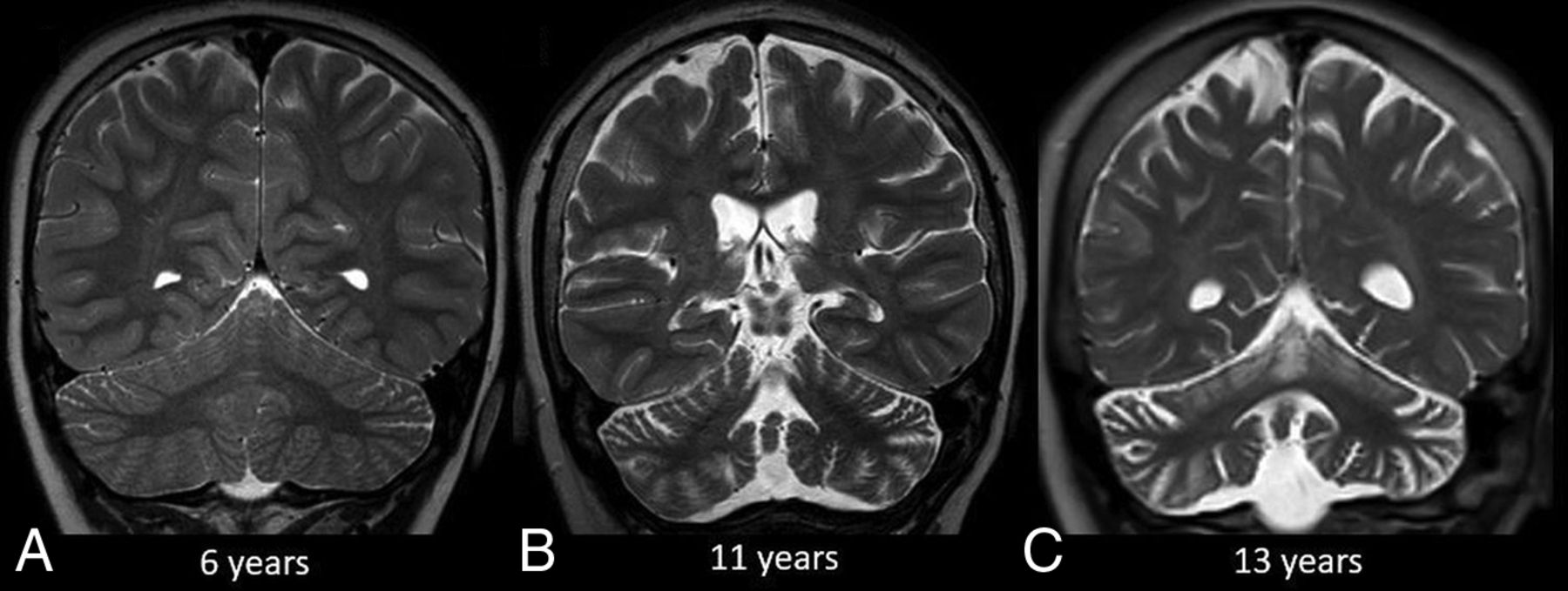
Sample cases of CLN2 disease are shown in Figs 2 and 3.

A 4-year-old boy with CLN2 disease. Axial FLAIR MR imaging shows hypointense thalami and hyperintense insular/subinsular region and posterior limb of the internal capsule (A); hyperintense periventricular and deep white matter (B), and hyperintense ventral pons (C). Note diffuse cerebral and cerebellar volume loss (A–C).

Progression of CLN2 disease. Coronal T2-weighted MR imaging at 6 years of age (A) shows no discernible volume loss. Follow-up MR imaging at 11 and 13 years of age (B and C) shows progressive cerebral and cerebellar volume loss. Note the greater degree of cerebellar volume loss relative to the cerebrum.
CLN3 Disease
In CLN3 disease (n = 2), the age of onset was 10 years in patient 3A and was unknown in patient 3B. MR imaging of patient 3A performed at 6 years of age showed no signal abnormality or volume loss on the initial MR imaging. Follow-up MR imaging at 10 years of age showed supratentorial volume loss with PVWM and DWM signal abnormality. MR imaging of patient 3B performed at 1.2 years of age showed only mild supratentorial atrophy without signal change and no obvious progression on follow-up imaging after 6 years (the first MR imaging for patient 3B was actually performed for follow-up of intraventricular hemorrhage; therefore, the atrophy was possibly not related to NCL). Neither patient had cerebellar atrophy.
CLN5 Disease
In CLN5 disease (n = 1), the disease onset was at 3.5 years of age. MR imaging at 6.5 years of age showed T2/FLAIR hypointense thalami; and T2/FLAIR hyperintensity in basal ganglia, PVWM, DWM, PLIC, I-SI region, and pons, along with mild supratentorial and severe cerebellar atrophy. There was no follow-up MR imaging.
CLN6 Disease
In CLN6 disease (n = 2), the age at disease onset was 4 years in a female and 9 years in a male patient. MR imaging of both patients (6A and 6B, at 8.6 years and 10.8 years of age) showed T2/FLAIR hypointense thalami, and both showed T2/FLAIR hyperintense signal in the PLIC, PVWM, DWM, and pons (none in the SCWM). One (patient 6A) had T2/FLAIR hyperintensity in the basal ganglia. Patient 6A showed severe supratentorial and cerebellar atrophy and brain stem atrophy. Patient 6B did not have atrophy on the initial MR imaging but showed mild supratentorial atrophy on follow-up imaging after 4.5 years. Both patients showed a greater degree of cerebral atrophy compared with cerebellar atrophy.
CLN7 Disease
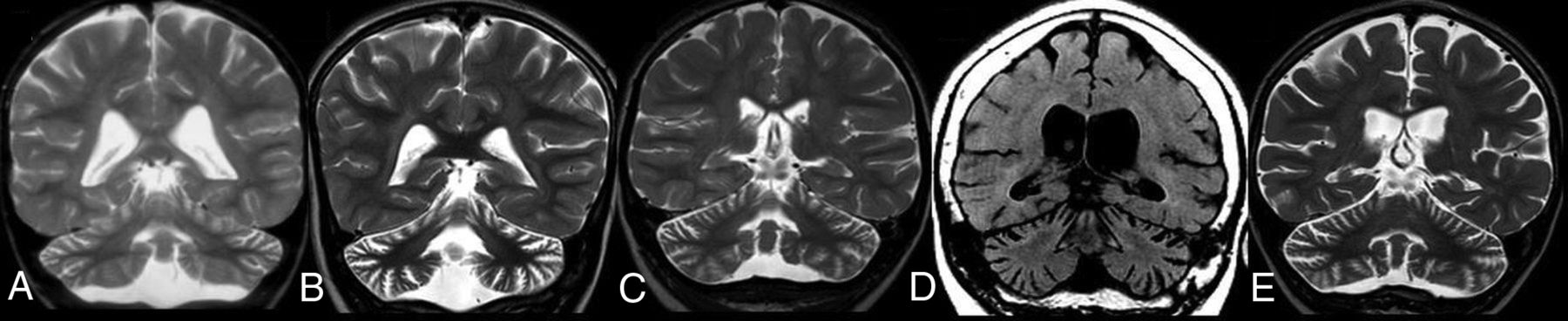
In CLN7 disease (n = 6), the median age of onset of disease was 3.75 years (IQR, 3.1 years), while the median age at first MR imaging was 4.5 years (IQR, 4 years). All patients showed T2/FLAIR hypointense thalami; 2 showed a T2/FLAIR hyperintense basal ganglia; all showed T2/FLAIR hyperintense PLIC, PVWM, and DWM (none in the SCWM) and 5 showed abnormal signal in the I-SI region and pons. Five patients showed supratentorial atrophy (mild = 3; moderate = 2), while all 6 patients showed cerebellar atrophy (mild = 1; moderate = 4; severe = 1). Four patients had brain stem atrophy. In 5 of 6 patients, the degree of cerebellar atrophy was greater than cerebral atrophy (Fig 4), with equal atrophy in 1 patient. None of the patients had follow-up MR imaging.

CLN7 disease. Coronal T2-weighted MR imaging of 4 different patients (A, B, C, and E) and coronal reformatted CT of 1 patient (D). Note cerebellar more than cerebral volume loss in all patients.
CLN8 Disease
In CLN8 disease (n = 2), the age of disease onset was 3.7 years in a male (8A) and 3.3 years in a female (8B) patient. MR imaging of both patients (at 4.5 and 3.3 years, respectively) showed T2/FLAIR hypointense thalami and T2/FLAIR hyperintense basal ganglia, PVWM, DWM, PLIC, I-SI region, and pons. None had SCWM signal abnormality. Both showed supratentorial (mild = 1; moderate = 1) and cerebellar (mild = 1, moderate = 1) atrophy. Patient 8A had greater cerebellar atrophy compared with cerebral atrophy while patient 8B had an equal degree of atrophy. Neither of the patients had follow-up MR imaging.
DISCUSSION
We report 24 patients with genetically confirmed NCL and their brain MR imaging features, which is one of the largest patient cohorts, to the best of our knowledge. Brain MR imaging findings of atrophy and white matter signal changes in NCL were first reported by Machen et al,33 expanding the findings on previously reported brain MR imaging features of NCL on CT.34 To the best of our knowledge, thalamic T2 hypointensity and periventricular T2 hyperintense rims in infantile-onset NCL were reported by Vanhanen et al35 for the first time in 1994. The white matter signal change was hypothesized to represent loss of myelin due to Wallerian degeneration based on histopathologic and microscopic studies.7,36-38 The thalamic T2 darkening was speculated to represent accumulation of iron, though this was debated.38 Vanhanen et al14 studied postmortem MR imaging with histopathologic correlation in 8 patients with infantile NCL in 1995 and demonstrated almost complete loss of cortical neurons, axons, and myelin sheaths in all patients, along with astrocytic proliferation containing periodic-acid-Schiff-positive storage material. They also demonstrated loss of thalamic neurons and loss of granular and Purkinje cells in the cerebellar cortex. They speculated that the thalamic T2 hypointense signal was due to accumulation of saposin (similar to GM2 gangliosides) and also by hypertrophic astrocytes containing storage protein and glial fibrillary acid protein.
In addition to their previous reports, Vanhanen et al,18 in 1995, reported progressive cerebral atrophy in infantile-onset NCL starting from 13 months of age and peaking at 4 years of age, redemonstrated by the same group in 2004 in patients with proved CLN1,20 with cerebellar atrophy lagging behind cerebral atrophy. All 6 of our patients with CLN1 disease (infantile onset) showed this pattern of initial early-onset cerebral atrophy followed by later-onset cerebellar atrophy.
Patients with late infantile-onset NCL have been reported to show early-onset cerebellar atrophy.23,39,40 We also identified similar findings in our late-infantile-onset CLN2, CLN5, and CLN7 diseases, wherein most patients showed earlier onset and, in most, a greater degree of cerebellar atrophy compared with cerebral atrophy. Interestingly, this was most remarkable in our CLN7 disease cohort wherein 5 of 6 patients showed this finding. On the other hand, both patients with CLN6 disease (early-juvenile and late-infantile NCL) had a greater degree of cerebral atrophy compared with cerebellar atrophy.
Juvenile NCL generally demonstrates cerebral and cerebellar atrophy, usually after the ages of 9 and 13 years respectively,19 though quantitative white matter signal intensities may be abnormal at a younger age.19 Studies have also demonstrated progressive hippocampal atrophy,25 altered white matter microstructure,22 and decreased volume of the dorsomedial thalami and corona radiata in CLN3 disease.24 One of our patients (3A) with CLN3 disease had mild PVWM and DWM signal abnormality and mild supratentorial volume loss at 14 years of age, while the other patient (3B) was only imaged up to 7 years of age and had no discernible signal abnormality. Unfortunately, we do not have morphometric data on these images.
White matter signal abnormalities, starting from the periventricular white matter with progression to deep and subcortical white matter in NCL, have been reported and have been attributed to white matter degeneration or abnormal myelin.18,19 We noted a distinctive abnormality in our cohort with involvement of the insular/subinsular region seen in 75% of our patients. To the best of our knowledge, this brain MR imaging feature has not been reported previously. Additionally, we identified T2/FLAIR hyperintensity of the PLIC and ventral pons in 91% and 79%, respectively, in our study cohort, which has been rarely reported in the literature.17,18 Jadav et al17 reported PLIC hyperintensity as an uncommon-but-characteristic brain MR imaging feature of CLN3 disease, though Holmberg et al23 also had previously reported this finding in a CLN5 disease cohort. In our study, this was seen not only in juvenile-onset but also in infantile and late-infantile onset NCL.
The differences in imaging phenotypes (eg, predominant cerebral over cerebellar atrophy and vice versa) is intriguing. As alluded to earlier, different subtypes of NCL may represent distinct disorders, each with its own molecular pathway converging to a common end point (including storage of autofluorescent material and neuronal death). The NCL genes encode several types of gene products that include lysosomal enzymes, soluble lysosomal proteins, transmembrane domain–containing proteins, and other proteins with different subcellular localizations.1,41 The precise localization and function of many of these proteins remain elusive. However, several roles of these proteins have now been elucidated, including apoptosis and autophagy, endocytosis, vesicular trafficking, cell proliferation, pH homeostasis, synaptic functioning, and protein secretion.1,41 Furthermore, researchers42 have demonstrated interaction among NCL proteins, with the CLN5 protein interacting with CLN2 and CLN3 polypeptides, suggesting the possibility of common pathways among these subtypes. In a recent study, CLN1, CLN3, CLN6, and CLN8 diseases have been linked to endoplasmic reticulum stress resulting in apoptosis.43 Whether the cerebral cortex is particularly susceptible to endoplasmic reticulum stress–related apoptosis accounting for predominant cerebral atrophy in these subtypes is unclear but appears plausible. The pathogenesis of NCL is therefore multifactorial and occurs not only due to the accumulation of lipopigments but also due to mitochondrial dysfunction, endoplasmic reticulum stress, and alterations in cellular pH, which lead to excessive production of mitochondrial reactive oxygen species and disturbed calcium homeostasis, resulting in apoptosis and neuronal loss. Each of the NCL proteins has varying effects on these cellular functions, resulting in slight differences in phenotypes and neuroimaging features.
This study has several limitations. First, because it was a retrospective study, MR imaging performed in different machines and protocols was analyzed, with nonuniform imaging parameters. Second, because images were analyzed during a 30-year period, there was an insufficient number of cases with the data required to perform meaningful quantitative analyses. We, therefore, restricted the study to qualitative analysis of signal intensity and volume loss. Third, only 9 patients had follow-up imaging, making it difficult to assess progression of disease. Last, there was under-representation of certain subtypes (eg, CLN3 disease) in this study cohort.
CONCLUSIONS
CLN1 and CLN7 diseases were the most common subgroups in our study cohort. The difference in the median time from disease onset and diagnosis was 1.5 years. Given recent FDA approval of the use of cerliponase alfa for treatment of CLN2 disease and ongoing phase I/IIa clinical trials with gene therapy for CLN3 and CLN6 diseases, it is imperative to reduce this time gap to arrest disease progression. In patients with a history of developmental regression, brain MR imaging and referral to a neurologist and geneticist are crucial to diagnose patients at the disease onset.
We identified reported classic neuroimaging features in all except 1 patient with NCL in our study, including predominant cerebellar-over-cerebral atrophy in CLN2, CLN5, and CLN7 diseases and minimal abnormality up to early adolescence in CLN3 disease. We confirmed less frequently reported findings of abnormal signal intensity in the deep white matter, posterior limb of the internal capsule, and ventral pons and demonstrated that these findings are more common than previously reported in the literature. We report abnormal signal intensity in the insular/subinsular region for the first time.
ACKNOWLEDGMENTS
A.B. is partially funded by Ontasian Imaging Laboratory (OIL), Toronto.
Footnotes
Disclosures: Saadet Mercimek-Andrews—UNRELATED: Board Membership: Recordati, BioMarin advisor; Consultancy: Canadian Medical Protective Association expert reviewer, Borden Ladner Gervais expert reviewer; Grants/Grants Pending: Physician Services Incorporation grant*; Payment for Lectures Including Service on Speakers Bureaus: BioMarin, Recordati. Manohar Shroff—RELATED: Other: BioMarin, Comments: I received a one-time honorarium (November 2019) for participating in an afternoon discussion on CLN2 hosted by BioMarin, which manufactures an enzyme-replacement treatment for CLN2. This was 2 months before we started this project. *Money paid to the institution.
Previously presented in poster format at: Annual Meeting of the American Society of Pediatric Neuroradiology, January 10–12, 2020; Miami Beach, Florida.
References
- Received May 2, 2020.
- Accepted after revision June 16, 2020.
- © 2020 by American Journal of Neuroradiology



{kind=link}
{kind=link}
{kind=link}
{kind=link}