
Article Figures & Data
Figures
- FIG 1.
This diagram highlights the chromosomal distribution of genes involved in the old (green boxes) and newly recognized (red boxes) CNS genetic tumor syndromes, with representation of 7 of the 8 new entities. The SDH gene implicated in familial paraganglioma is located along the inner membrane of mitochondria and is not depicted here. VHL indicates Von Hippel-Lindau syndrome.
- FIG 2.
ELP1-related medulloblastomas in siblings. A 6-year-old boy (sibling A) with MR imaging revealing a heterogeneously T2 hyperintense (A, axial T2 TSE) mass with cystic changes and heterogeneous enhancement (B, coronal postcontrast). Histopathology with immunohistochemistry revealed a large-cell anaplastic-pattern WHO grade 4, SHH-activated (TP53 wild-type) medulloblastoma. The patient’s older brother, an 18-year-old boy (sibling B), was diagnosed with a similar tumor 7 years later. MR imaging revealed a right lateral cerebellar T2 hyperintense mass (C, axial T2 TSE) with solid contrast enhancement (D, coronal postcontrast). Pathology revealed an SHH-activated (TP53 wild-type) medulloblastoma with desmoplastic/nodular morphology. A neuro-oncology genetic panel (utilizing NGS technique) in both siblings showed biallelic inactivation of ELP1 due to somatic loss of chromosome arm 9q along with homozygous deletion of the tumor-suppressor gene PTCH1, confirming an ELP1 germline mutation. Both siblings remain disease-free (9 years postsurgery for sibling A, and 1 year for sibling B) (Online Supplemental Data).
- FIG 3.
Rhabdoid intracranial and spinal meningiomas with loss of BAP1 expression. Left lateral cerebellomedullary cistern enhancing mass (A, axial postcontrast T1, white arrow) in a 36-year-old man, extending into the hypoglossal canal, revealing atypical meningioma (CNS WHO grade 2) on histology, with rhabdoid features (B) and loss of BAP1 expression on immunohistochemistry (Online Supplemental Data). Coronal (C) and axial (D) contrast-enhanced MR images in a different patient depict an extramedullary cervical mass extending along the left neural foramen (arrows). Histopathology (not shown) revealed rhabdoid meningioma with loss of BAP1 expression.
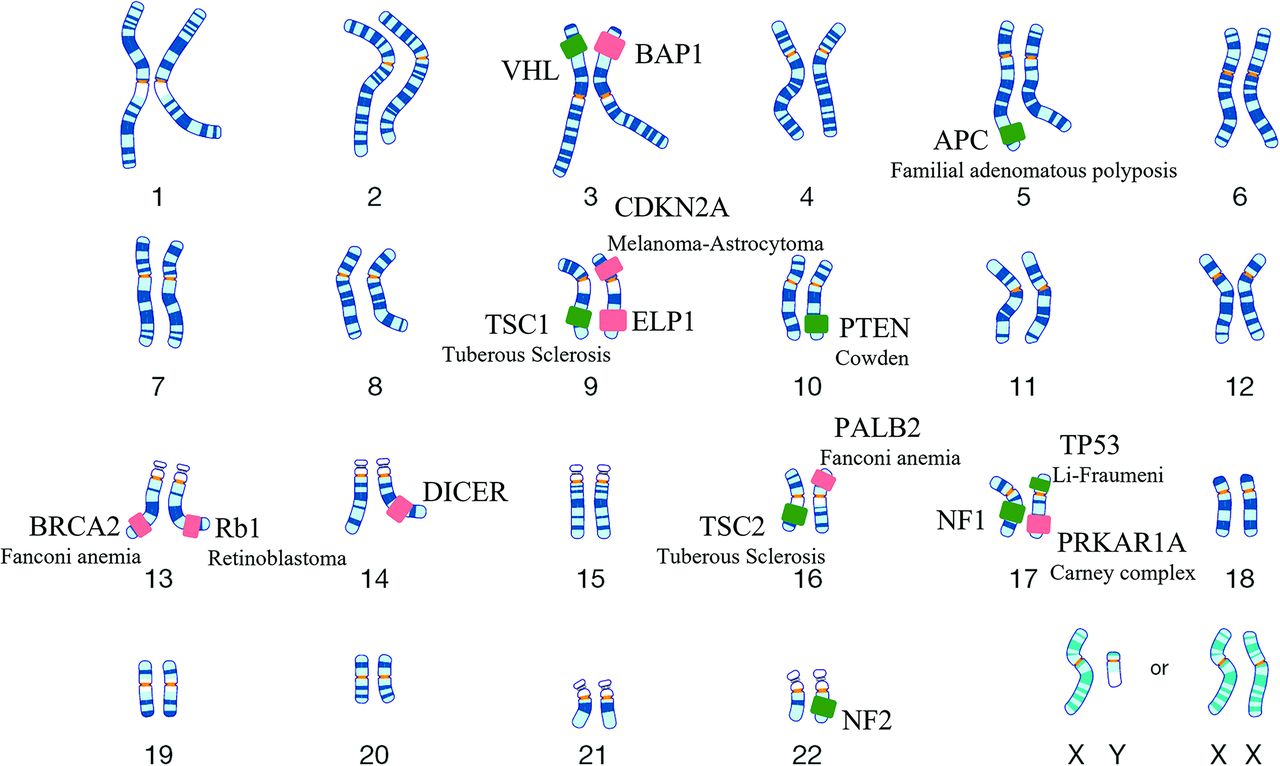
- FIG 4.
DICER1-mutant intracranial sarcoma. Sagittal T2 (A) and postcontrast (B) MR images in a 16-year-old adolescent girl reveal a dural-based left parasagittal T2-hyperintense enhancing mass, presumed to be a meningioma on imaging. Histopathology, however, revealed spindle and pleomorphic cells (C), and immunohistochemistry tests were negative for GFAP, OLIG2, SSTR2 (D), STAT6, S-100, and SOX10, arguing against glial, meningothelial, or melanocytic tumors. A Somatic Disease/Germline Comparator Exome sequencing panel revealed a pathogenic germline DICER1 mutation. Overall, the histomorphologic and immunophenotypic findings supported a sarcomatoid neoplasm. The additional presence of pathogenic DICER1 mutations was diagnostic of primary intracranial sarcoma, DICER1-mutant. Because the prognosis for patients with DICER1-mutant primary intracranial sarcoma remains unknown due to limited clinical data, a CNS WHO grade designation was not rendered, according to the 2021 WHO Classification of Tumors of the Central Nervous System.
- FIG 5.
MMNST in a 26-year-old woman with CNC. Coronal MR images (A and B) reveal a mass along the left S1–S2 neural foramen with intrinsic T1-hyperintense content (A, T1-weighted, non-fat-saturated, arrow) with peripheral enhancement and a central nonenhancing component (B, postcontrast with fat saturation, arrow). Note marked expansion of the neural foramen and erosive osseous changes along the inferior edge on coronal CT (C, arrow). Histopathology reveals a spindle cell (D, white arrow) neoplasm with psammomatous bodies (D, black arrow). Immunohistochemistry was positive for SOX10, S-100, and MelanA, confirming the diagnosis of MMNST. Neoplastic cells showed complete loss of PRKAR1A, confirming an association with CNC (Online Supplemental Data).
- FIG 6.
HNPGs in 2 different patients, one with SDH mutation (patient A) and the other with normal SDH expression (patient B). Patient A has familial paraganglioma syndrome with multiple HNPGs and a left adrenal phaeochromocytoma (A, arrowheads) on a 68Ga-DOTATATE PET scan, with complete loss of SDH expression on immunohistochemistry (C). A solitary HNPG along the right carotid artery is noted on a 68Ga-DOTATATE PET scan in patient B (B, arrow) with normal SDH expression on pathology (D).
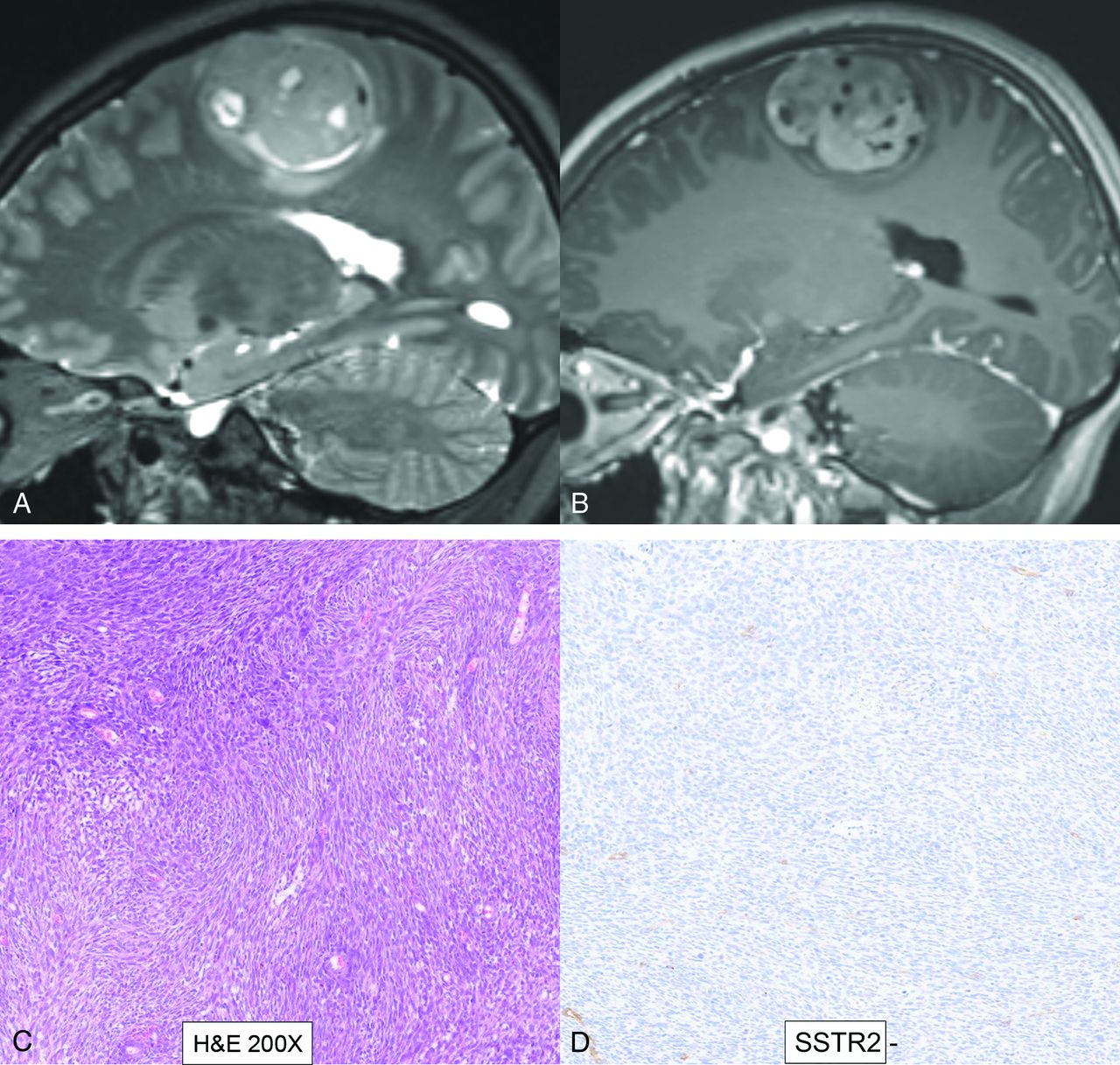
- FIG 7.
Cerebroretinal vasculopathy and leukoencephalopathy in a 20-year-old man with FA. MR imaging including a sagittal T2-weighted image (A), axial diffusion (B), T2 TSE (C), and postcontrast (D) images reveal a large area of white matter edema with heterogeneous enhancement and central necrosis in the right frontal lobe. The primary radiographic differential included a high-grade glial neoplasm, and a biopsy was performed. Histopathology revealed severe vasculopathy exhibiting vascular hyaline changes and perivascular and transmural chronic inflammation. The background white matter demonstrated extensive vacuolization and gliosis with necrosis surrounding the vessels. No evidence of JC virus or Epstein-Barr virus was identified by in situ hybridization, and no fungal organisms were detected. Follow-up MR imaging (Online Supplemental Data) after 6 months of immunosuppressive treatment showed marked reduction in the right frontal edema and enhancement, however, with new areas of edema and enhancement in the left parieto-occipital lobe. Advanced veno-oclusive retinopathy with neovascularization was noted on fundoscopy (Online Supplemental Data).







Tables
Newly recognized CNS genetic tumor syndromes in the 5th WHO classification with updates on genetic pathway
WHO Classification, 4th Edition 2016 WHO Classification, 5th Edition 2022 Familial Tumor Syndromes (Chapter 16) Genetic Tumor Syndromes Involving the CNS (Chapter 14) New Syndromes Genetic Pathway Most Common Nervous System Tumors NF type 1 ELP1-medulloblastoma Growth factor receptor Medulloblastoma NF type 2 BAP1 tumor predisposition Ubiquitin protein Meningioma Schwannomatosis DICER1 syndrome microRNA regulation Metastasis (from pulmonary blastoma) Von Hippel-Lindau syndrome CNC PKA signaling pathway Malignant melanotic Nerve sheath tumor Tuberous sclerosis Melanoma-astrocytoma Cell cycle and apoptosis Glioma Li-Fraumeni syndrome Familial paraganglioma Oxidative stress (Krebs) Paraganglioma Cowden syndrome FA DNA repair and genomic stability Medulloblastoma Turcot syndrome Familial retinoblastoma Cell cycle and apoptosis Retinoblastoma Constitutional mismatch repair deficiency syndrome Familial adenomatous polyposis 1 All entities from 4th edition kept in the new edition, except Turcot syndrome Nevoid basal cell carcinoma syndrome Constitutional mismatch repair deficiency (previously subtype under Turcot syndrome) Rhabdoid tumor syndrome Familial adenomatous polyposis 1 (previously subtype under Turcot syndrome)


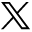


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.