
Abstract
BACKGROUND AND PURPOSE: Temporal bone imaging plays an important role in the work-up of branchio-oto-renal syndrome. Previous reports have suggested that the unwound or offset cochlea is a highly characteristic marker for branchio-oto-renal syndrome. Our goals were to examine the prevalence of this finding in a branchio-oto-renal syndrome cohort and analyze genetic-phenotypic associations not previously established.
MATERIALS AND METHODS: This multicenter retrospective study included 38 ears in 19 unrelated individuals with clinically diagnosed branchio-oto-renal syndrome and confirmed mutations in the EYA1 or SIX1 genes. Two blinded neuroradiologists independently reviewed and documented temporal bone imaging findings in 13 categories for each ear. Imaging phenotypes were correlated with genotypes.
RESULTS: There was excellent interrater agreement for all 13 phenotypic categories (κ ≥ 0.80). Of these, 9 categories showed statistically significant differences between patients with EYA1-branchio-oto-renal syndrome and SIX1-branchio-oto-renal syndrome. Cochlear offset was present in 100% of patients with EYA1-branchio-oto-renal syndrome, but in only 1 ear (12.5%) among patients with SIX1-branchio-oto-renal syndrome. A short thorny appearance of the cochlear apical turn was observed in most patients with SIX1-branchio-oto-renal syndrome.
CONCLUSIONS: An offset cochlea is associated with the EYA1-branchio-oto-renal syndrome genotype. The SIX1-branchio-oto-renal syndrome genotype is associated with a different cochlear phenotype that almost always is without offset and has a short thorny tip as the apical turn. Therefore, cochlear offset is not a characteristic marker for all patients with branchio-oto-renal syndrome. The lack of a cochlear offset in a patient with clinically suspected branchio-oto-renal syndrome does not exclude the diagnosis and, in fact, may be predictive of the SIX1 genotype.
ABBREVIATIONS:
- BOR
- branchio-oto-renal syndrome
- ET
- Eustachian tube
- IAC
- internal auditory canal
- LSCC
- lateral semicircular canal
- PSCC
- posterior semicircular canal
- VA
- vestibular aqueduct
Branchio-oto-renal syndrome (BOR) is a common cause of congenital hearing loss, occurring in about 2% of profoundly deaf children,1,2 with a prevalence estimated at 1 in 40,000.1
Melnick et al3 first described autosomally inherited familial branchio-oto-renal dysplasia in a family with hearing loss, cochlear malformation, malformed pinnae, prehelical pits, branchial cleft fistulas, and renal dysplasia. Subsequent authors found families having similar features but variability in renal abnormalities (branchio-oto or branchio-oto-ureteral syndrome);1,2,4 these were attributed to variable gene penetrance.2,5 In 1980, Fraser et al1 observed that not all patients with BOR had hearing loss. Anatomic studies described abnormal ossicles, hypoplastic cochleae, dysplastic semicircular canals, enlarged vestibular aqueducts (VAs), abnormal facial nerves, and bulbous internal auditory canals (IACs).6⇓-8 Current diagnostic criteria are clinically based and dependent on the presence of major and minor criteria (Table),9 including deafness and anatomic malformations of the ear, neck, and kidney as previously described.
Diagnostic criteria for BOR syndrome (Chang et al9)a
Mutations in EYA1 were first reported in 1997 as a genetic cause of BOR.10 Subsequently, mutations in SIX1 were also identified in individuals with BOR. Approximately 40% of patients with BOR have mutations in the EYA1 gene9 and <4% have mutations in the SIX1 gene.11 EYA1 and SIX1 are critical to mammalian organogenesis, including the otic vesicle. SIX1 is a transcription factor and is reliant on the binding of EYA-family proteins at the conserved N-terminal (SIX domain) for transcription activation.12,13
Large cohort studies have shown causative genetic mutations identified in only 36%–72% of clinically diagnosed individuals with BOR;14⇓-16 therefore, evaluating the phenotype remains essential for diagnosis, even as genetic testing plays an increasingly important role.9,14,17⇓⇓-20
Various temporal bone abnormalities have been described in BOR affecting the ossicles, inner ear, facial nerve course, and IAC. Cochlear malformation is common and is described as anteromedial offset of hypoplastic middle/apical turns away from a tapered basal turn.5,21⇓-23 This morphologic appearance has been termed “unwound” or “offset” and has been reported to be a highly specific marker for BOR.23,24
In this study, we focused on BOR genotype-phenotype associations on imaging that have not been previously established. Specifically, we examined the prevalence of the offset cochlea among patients with BOR with a known causative genotype, determined whether there are other temporal bone differences between patients with EYA1-BOR and SIX1-BOR, and described a new morphologic feature of the SIX1-BOR cochlea.
MATERIALS AND METHODS
Patients
This was a multicenter, retrospective review of cases of genetically confirmed BOR syndrome. Patients from 4 institutions were recruited; inclusion criteria were the following: clinical diagnosis of BOR syndrome referred from specialties including Otology and General/Pediatric Otorhinolaryngology; known causative genetic mutation associated with BOR; and diagnostic temporal bone CT and/or MR imaging available for review. This study was approved by the institutional review board/ethics committee of each institution.
Imaging Methods
All CT scanners across the participating institutions were helical multidetector CT scanners with parameters as follows: 120 kV(peak), 100–200 mA, section thickness = 0.6–0.625 mm. All MR imaging scanners across the participating institutions were 3T, with the sequence assessed being heavily T2-weighted (driven equilibrium radiofrequency reset pulse [DRIVE; Philips], constructive interference in steady state [CISS; Siemens], or T2 sampling perfection with application-optimized contrasts by using different flip angle evolution [SPACE sequence; Siemens] depending on the vendor). Axial reformats of the temporal bones were created for both CT and MR imaging studies in a plane parallel to the lateral semicircular canal (LSCC) (or estimated as such in cases of anomalous LSCC) and coronal planes perpendicular to the true axial planes before image analyses.
Image Review
Two neuroradiologists with 5 (J.P.) and 15 (A.F.J.) years of experience reviewed all 19 cases. The reviewers were blinded to the original reports, demographics, genetic diagnosis, and the other reviewer’s findings. For each case, the following 13 parameters were assessed on the basis of existing definitions and/or descriptions in the literature:22,24⇓⇓⇓⇓⇓⇓⇓⇓-33
Cochlear offset: yes/no
Cochlear hypoplasia: yes/no (hypoplasia defined as ≤4.3 mm in height in the coronal plane26,27)
Apical hypoplasia: yes/no; morphology if present
Modiolus: normal/abnormal/absent
Cochlear fossette stenosis: <1.4 mm/1.4–1.8 mm/>1.8 mm (fossette width measured in the axial plane spanning the inner margins of the bony edges across the base of the modiolus; stenosis was defined as <1.4 mm [in accordance with lower limits in the literature to avoid false-positives]; borderline as 1.4–1.8 mm; and not stenotic as >1.8 mm30⇓⇓-33)
LSCC bone island small: yes/no (normal defined as >3 mm27)
Posterior semicircular canal (PSCC) anomalous configuration: yes/no
VA enlargement: yes/no (when equivocal, Pöschl views were generated, and >0.9 mm at midpoint was considered enlarged29)
Facial nerve canal course medially deviated: yes/no
Facial nerve canal widened: yes/no (widened defined as >1.42 mm22)
IAC widened (bulbous- or funnel-shaped): yes/no
Eustachian tube (ET) dilated: yes/no
Ossicular anomalies: yes/no
To accurately assess cochlear morphology, including whether there was apical hypoplasia, we took care to count the turns of the cochlea correctly. Fitch et al7 and Chen et al5 mentioned “fifths” of a cochlea, with an absent apical turn resulting in four-fifths of the cochlea remaining, designating each half turn as one-fifth of a cochlea, for a total of 2.5 turns. By means of this method, the basal turn spans the length from the round window to the medial bend (the first fifth) and then from the medial bend back laterally (second fifth), completing the basal turn. The middle turn then starts from lateral to medial (the third fifth) and then from medial back to lateral (fourth fifth). Finally, the apical turn extends from lateral to medial (last fifth) (Online Supplemental Data). The portion of the cochlea close to the round window has a bulbous configuration that contributes to the outward convex shape of the cochlear promontory. It contains the most basal part of the cochlear duct and the cul-de-sac of the endolymphatic space where the osseous spiral lamina, spiral ligament, and basilar membrane merge. This portion of the cochlea forms a “three-dimensional (3D) ‘fish-hook’-like shape”34⇓-36 and has been referred to as the “hook region” (Online Supplemental Data).37,38 The hook region extends from the edge of the vestibule and round window to the point where the cochlea begins to coil. For consistency during image assessment, we considered the hook region of the cochlea as part of the basal turn and thus part of the first fifth.
When there was a disagreement between the 2 reviewers, consensus reading (blinded to genotype) was performed.
Statistical Analysis
Comparison among group demographics was made with the Mann-Whitney U test for continuous nonparametric variables. A Cohen weighted κ statistic was calculated to assess interrater reliability for each phenotypic parameter and categorized as poor (<0.2), fair (0.20–0.39), moderate (0.40–0.59), good (0.60–0.79), or excellent (≥0.80). Differences between the EYA1-BOR and SIX1-BOR groups for each parameter were assessed using the Fisher exact test. P = .05 was set as the threshold of significance. Statistical analyses were performed using SAS Studio, Version 3.8 (SAS Institute).
RESULTS
Nineteen unrelated individuals (38 ears) met the inclusion criteria; 15 individuals (30 ears) had mutations in the EYA1 gene, and 4 individuals (8 ears) had mutations in the SIX1 gene.
Of the 19 patients, 14 underwent CT and 5 underwent MR imaging. Among the EYA1 genotype population, there were 11 males and 4 females, with a median age of 4.5 years (range, 2 months to 26 years). Among the SIX1 genotype population, there were 3 males and 1 female, with a median age of 4 years (range, 3 months to 13 years). There were no statistically significant differences in age and sex among the groups.
Thirteen phenotypic parameters were examined for each ear. Results recorded by the 2 raters showed excellent interrater reliability (κ ≥ 0.80). A few discrepancies were limited to qualitative assessments, including determination of modiolar structure, VA enlargement, facial nerve canal course, IAC width, and ET dilation. Notably, perfect observer agreement was seen for the quantifiable phenotypes, including assessment of the cochlear offset.
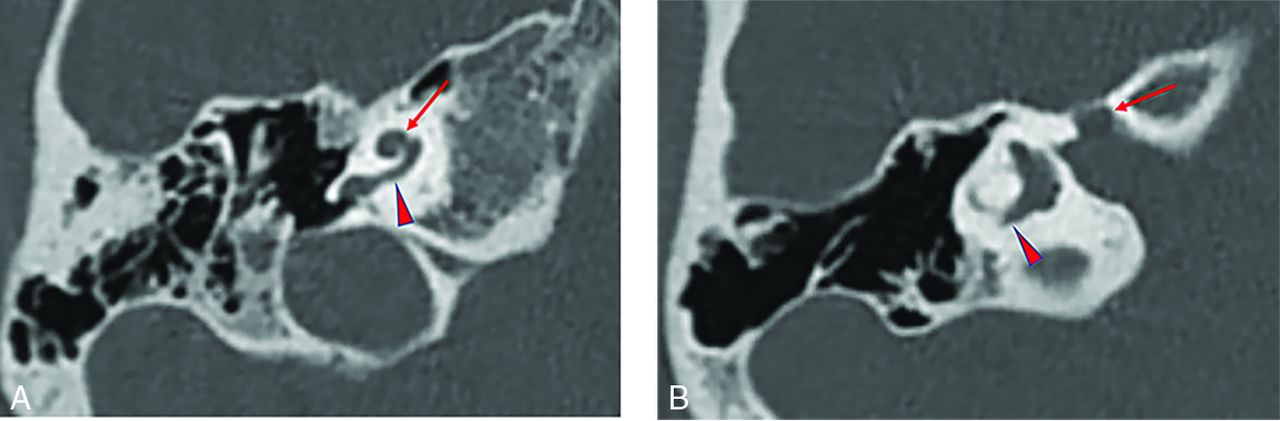
Differences in temporal bone features between patients with EYA1-BOR and SIX1-BOR reached statistical significance (P < .05) in 9 of 13 structures evaluated: cochlear offset, cochlear hypoplasia, apical turn hypoplasia, modiolar abnormality, VA enlargement, labyrinthine facial nerve canal medialization, IAC widening, ET dilation, and ossicular abnormalities (Online Supplemental Data). Of these, the structural feature that was most divergent between the 2 patient populations was cochlear offset, which was present in 100% of patients with EYA1-BOR and in only 1 ear among the patients with SIX1-BOR. This SIX1-BOR ear had an offset cochlea that did not resemble the offset cochleae seen among the EYA1-BOR population, including absence of basal turn tapering. The EYA1-BOR phenotype in our sample population was characterized by having cochlear offset, cochlear hypoplasia, apical turn hypoplasia, abnormal modiolus, VA enlargement, medialization of the facial nerve course, widening of the IAC, ET dilation, and ossicular anomalies (Fig 1). Individuals with SIX1 mutations were significantly less likely to have these phenotypic anomalies (Fig 2). Cochlear fossette stenosis, a small LSCC bone island, PSCC anomaly, and facial nerve canal widening were less prevalent in the SIX1-BOR group but did not reach a statistically significant difference compared with the EYA1-BOR group (P > .05). The Online Supplemental Data summarize the findings.

Temporal bone imaging appearance in a patient with EYA1-BOR. A, Axial CT image in bone algorithm shows an offset cochlea; the middle turn (arrow) is aligned more anteromedially than usual relative to the basal turn (arrowhead). The cochlea is, overall, small with a hypoplastic apical turn. B, Axial CT image in bone algorithm at a more superior level shows a medialized and widened labyrinthine facial nerve canal (arrow). The PSCC is anomalous, appearing as a blind-ending tubular structure (arrowhead).

Temporal bone imaging appearance in a patient with SIX1-BOR. A, Axial CT image in bone algorithm shows absence of the cochlear offset; the middle turn (long arrow) is normally aligned relative to the basal turn (arrowhead) without anteromedial displacement. The thorny apical turn can also be appreciated (short arrow). B, Axial CT image in bone algorithm at a more superior level shows a normal course of the labyrinthine facial nerve canal (arrow) without medialization or widening.
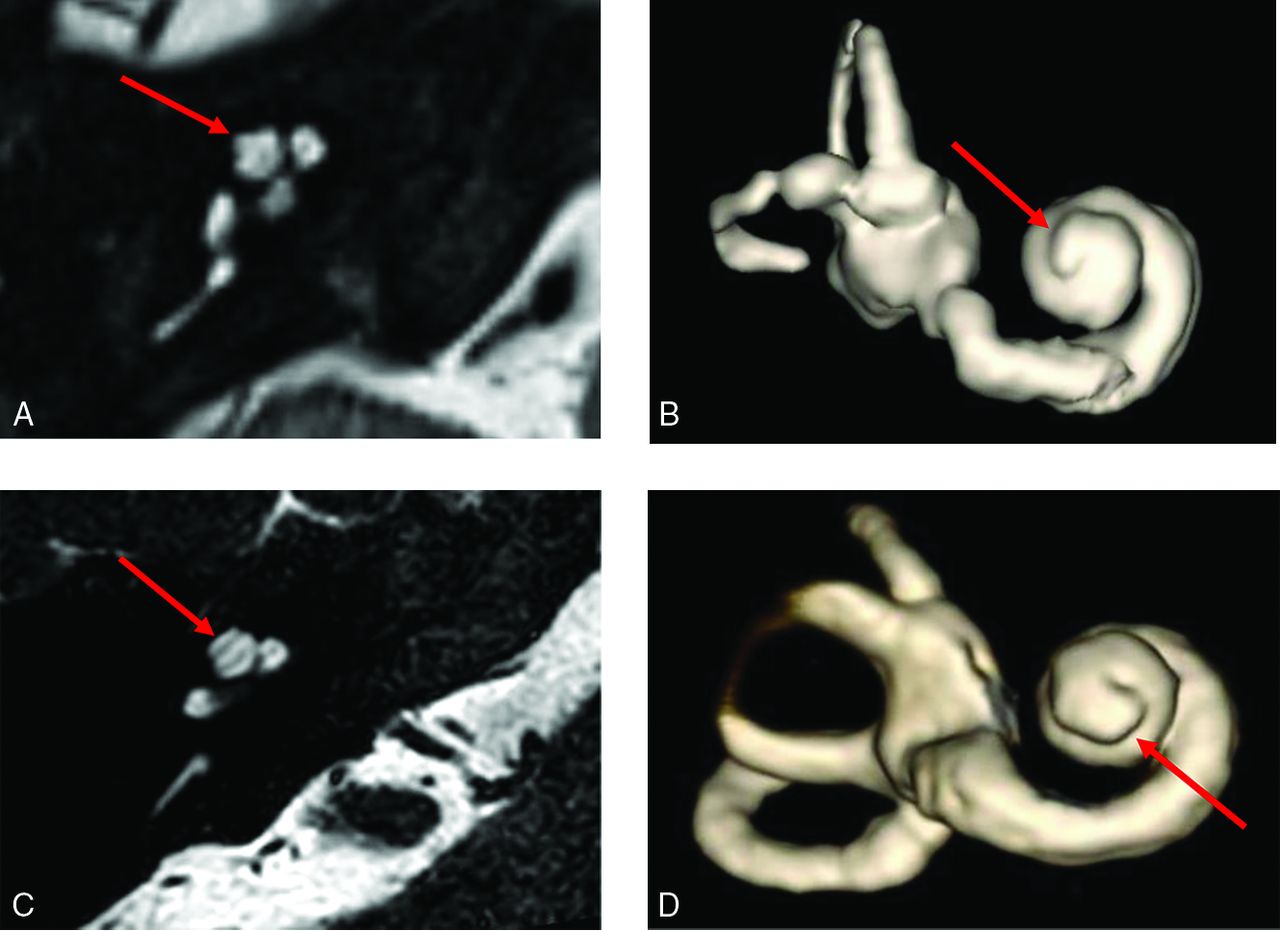
Among the SIX1-BOR group, the 1 ear with cochlear offset did not have an apical turn, and 2 ears (in 1 patient) did not allow detailed apical turn morphology assessment due to the spatial resolution of MR imaging in that study. In the remaining 5 ears, all had a distinct short protuberant appearance to the cochlear apical turn reminiscent of a poking thorn, which we termed a “thorny” apical turn (Fig 3).

Thorny apical turn of the SIX1-BOR cochlea. Axial heavily T2-weighted MR image (A) and 3D reconstruction of the cochlea (B) show a short, thin, protuberant projection forming the apical turn, imparting upon it a thorny morphology (arrows in A and B). Contrast it with the normal apical turn morphology in a patient with normal hearing and no history of BOR or sensorineural hearing loss (C and D), which is longer with a smooth flat appearance and rounded end (arrows in C and D).
DISCUSSION
In our assessment of 19 patients with BOR, the classic unwound or offset cochlea was seen among all those with the EYA1 mutation but in only 1 ear among those with the SIX1 mutation. Notably, however, this cochlea did not have the typical appearance of the EYA1-BOR offset cochlea with a tapered basal turn but rather had features more closely resembling a cochlear hypoplasia type 4 (CH4) anomaly.39 In addition, the SIX1 genotype did not demonstrate most of the other previously described cochlear anomalies nor additional temporal bone findings such as VA enlargement, medially displaced labyrinthine facial nerve, widened IAC, ET dilation, and ossicular anomalies.5,21,22,40 Furthermore, most with the SIX1-BOR genotype had a characteristic short protuberant thorny apical turn, a feature not seen among the EYA1-BOR genotype population. This finding suggests distinct genotype-phenotype differences within BOR syndrome with regard to the temporal bone.
EYA1 was the first gene identified to cause BOR syndrome.41⇓⇓-44 It plays a role in the development of the inner ear and surrounding mesenchyme, as well as metanephric cells surrounding ureteric branches in renal development.45 In 2004, Chang et al9 analyzed phenotypic data from families with EYA1 mutations and proposed criteria for clinical diagnosis, ushering in the modern diagnostic criteria for BOR (Table). In 2005, Propst et al22 described temporal bone CT findings in 13 families with BOR syndrome diagnosed on the basis of the criteria of Chang et al. In 2006, the term “offset” was coined by Robson23 to describe “tapering of the basal turn of the cochlea,” and “small middle and apical turns that are offset anteriorly and appear separated from the basal turn” and deemed “characteristic of BOR syndrome.” Of note, many of these descriptions were made when EYA1 was the sole known gene for BOR syndrome.
However, in 2003 and 2004, Ruf et al17,18 identified a second gene locus that mapped to human chromosome 14q23.1, where the SIX1, SIX4, and SIX6 genes reside. Three different SIX1 mutations were identified in 4 BOR/branchio-oto (BO) kindreds. Thus, SIX1 became a second known gene that can be associated with BOR syndrome. Sanggaard et al19 discovered a low frequency (25%) of branchial arch malformation and the absence of renal pathology among patients with BOR with SIX1 mutations, and there was one patient with no temporal bone malformation. They suggested that there may be clinical differences in patients with BOR with EYA1 and SIX1 mutations. Genotype-phenotype correlation studies are, therefore, of much interest.
We discovered different temporal bone phenotypes: The SIX1 genotype did not show the classic temporal bone anomalies described in the radiology literature for BOR syndrome. In particular, the unwound or offset cochlea was observed in all individuals in our cohort with the EYA1 genotype but in almost none with the SIX1 genotype, showing instead a short, protuberant, thorny apical turn.
Mammalian inner ear development is a complex process dependent on the interaction of many genes. Studies in mouse models showed that during embryologic development, EYA1 and SIX1 are both expressed in the ventral part of the otic vesicle from which the cochlear structures are derived.46 Most important, expression of SIX1 in the otic vesicle is dependent on EYA1, whereas the expression of EYA1 is unaffected in SIX1 murine knockout models.47 This finding may explain why the cochlear phenotype in patients with EYA1-BOR is more severe than in patients with SIX1-BOR. It is of interest that the expression of SIX1 in the mammalian inner ear is more prominent in the apex of the cochlea than in the base.47 This feature may have relevance to the finding of a thorny apical turn in our cohort with the SIX1 genotype, but further work is needed to understand this issue.
Regarding the 1 case with a unilateral cochlear abnormality with features of CH4, it is unclear whether these features are within the wider spectrum of the SIX1 phenotype or if there is a secondary explanation. The SIX1 variant in this case (Cys16Tyr) is notable for its location toward the N-terminal (SIX domain) within an α helix critical for EYA interaction. Although this variant has not been reported before in the literature, a variant at the adjacent amino acid residue (V17E/Val17Glu) has been the subject of functional work. Patrick et al12,48 studied the effect of a number of SIX1 variants on the EYA-SIX1-DNA complex and concluded that SIX1-BOR mutations contributed to the disease pathology through at least 2 different mechanisms: In most cases (all C-terminal to this mutation and not in the α helix), the mechanism appeared to be related to diminished ability of SIX1 to bind DNA; another mechanism was seen in a case of the V17E mutation, in which formation of the SIX1-EYA complex was completely abolished and nuclear localization of the complex was not observed. This mechanism was not seen in any of the other SIX1-BOR mutations modeled.
Therefore, one theory to account for the unilateral cochlear abnormality in our SIX1 Cys16Tyr variant is that mutations in the α helical domain are functionally distinct and may lead to a different SIX1 inner ear phenotype. Of note, an unaffected sibling (SIX1 genetic analysis normal) of this proband had several ear tags, so it may be that there are secondary genetic modifying factors impacting the clinical presentation; EYA1 was also fully sequenced, and multiplex ligation-dependent probe amplification was undertaken in the proband with negative findings. Unfortunately, clinical data reported for the V17E case were limited and did not include imaging,11 and the number of reported SIX1-BOR cases in the literature overall remains small. It is difficult to draw any firm conclusions from this specific case, but functional studies of the Cys16Tyr SIX1 variant and further imaging in SIX1 cases, particularly those involving the α helix, may help resolve this issue.
Some limitations of this study include its retrospective nature, different CT/MR imaging protocols, and its relatively small sample size. In the future, we would benefit from a prospective clinicoradiologic study involving all organ systems involved in BOR syndrome for precise genotype-phenotype correlation.
CONCLUSIONS
We found a significant difference between the BOR EYA1 and SIX1 genotype-phenotypes in the temporal bone. The SIX1 phenotype is associated with a lack of cochlear offset, a thorny apical turn, as well as a relative absence of many other previously reported temporal bone anomalies. This shows that cochlear offset is not a characteristic feature nor a reliable indicator for all BOR cases; specifically, it is seen with the BOR EYA1 genotype but is not sensitive nor specific for the detection of the BOR SIX1 genotype. Thus, the lack of cochlear offset on imaging does not exclude the diagnosis of BOR and, in fact, may be predictive of the SIX1 genotype in an individual with clinically suspected BOR. Careful radiologic delineation of the temporal bone and inner ear structures has the potential to help establish specific and sensitive features to aid in multidisciplinary genomic variant interpretation in suspected cases of BOR.
Acknowledgments
We would like to thank Julia Wei, MPH, for statistical consultation, and Stephen Kovach, RT, for assistance with 3D modeling and construction.
Footnotes
J. Pao and F. D’Arco are co-first authors.
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
References
- Received August 1, 2021.
- Accepted after revision November 2, 2021.
- © 2022 by American Journal of Neuroradiology




{kind=link}
{kind=link}
{kind=link}