
In the past, hemodynamic influences were thought to primarily control the identity of arteries and veins. Today, it is accepted that blood vessel identity is determined before the onset of blood flow because vascular precursor cells express specific genetic markers. In addition to these molecular markers interacting in complex signaling pathways, there are epigenetic mechanisms that help determine and maintain endothelial cell fate. Here, we will attempt to give the readers some insight with regard to these new concepts. We also would like to speculate as to how these new discoveries may relate to neuroradiology, and our related thoughts are found in italics.
In the embryo, the cardiovascular system forms first.1 Formation of all other organs depends on oxygen delivery. Blood vessels form before blood starts flowing, and the circulatory system must be in place before organs develop. The oxygenated blood pumped into the arteries by the heart plays a role in the development of smooth muscle cells and an extracellular matrix capable of supporting high pressures. Once blood passes through capillaries, intraluminal pressure decreases and venous valves ensure unidirectional flow. These physiologic and mechanical changes were thought to be responsible for development of blood vessel identity—that is, flow direction, oxygen concentration, and pressure determine if vessels become arteries or veins. Recent investigations highlight the fascinating concept that endothelial cells are predestined, even before blood begins circulating, to become arteries or veins.
In embryonic life, angioblasts (endothelial precursor cells) arising mostly from mesoderm form epithelial tubes by apposition of cords.1 Creation of these tubes is called “vasculogenesis” and is a temporary and rapidly concluding prenatal mechanism.1–3 A second process, called “angiogenesis” is responsible for the formation of blood vessels from pre-existing ones (involving elongation, sprouting, and remodeling of those pre-existing blood vessels). Vasculogenesis leads to formation of the first blood vessels: the aorta and posterior cardinal vein. In some fish and mice, progenitor cells arising from the lateral mesodermal plates (alongside the notochord) give rise to cells that migrate to the midline under the notochord and establish the intermediate cell mass that subsequently assembles the dorsal aorta and cardinal vein.3,4 This early process is evidence of the already defined identity of endothelial cells. The genesis of arteries and veins from closely located progenitor cells is recapitulated throughout the entire body, explaining why arteries and veins are nearly always side by side. Even before progenitor cells begin migrating ventrally, their fate as either arterial or venous has been cast. How does this happen?
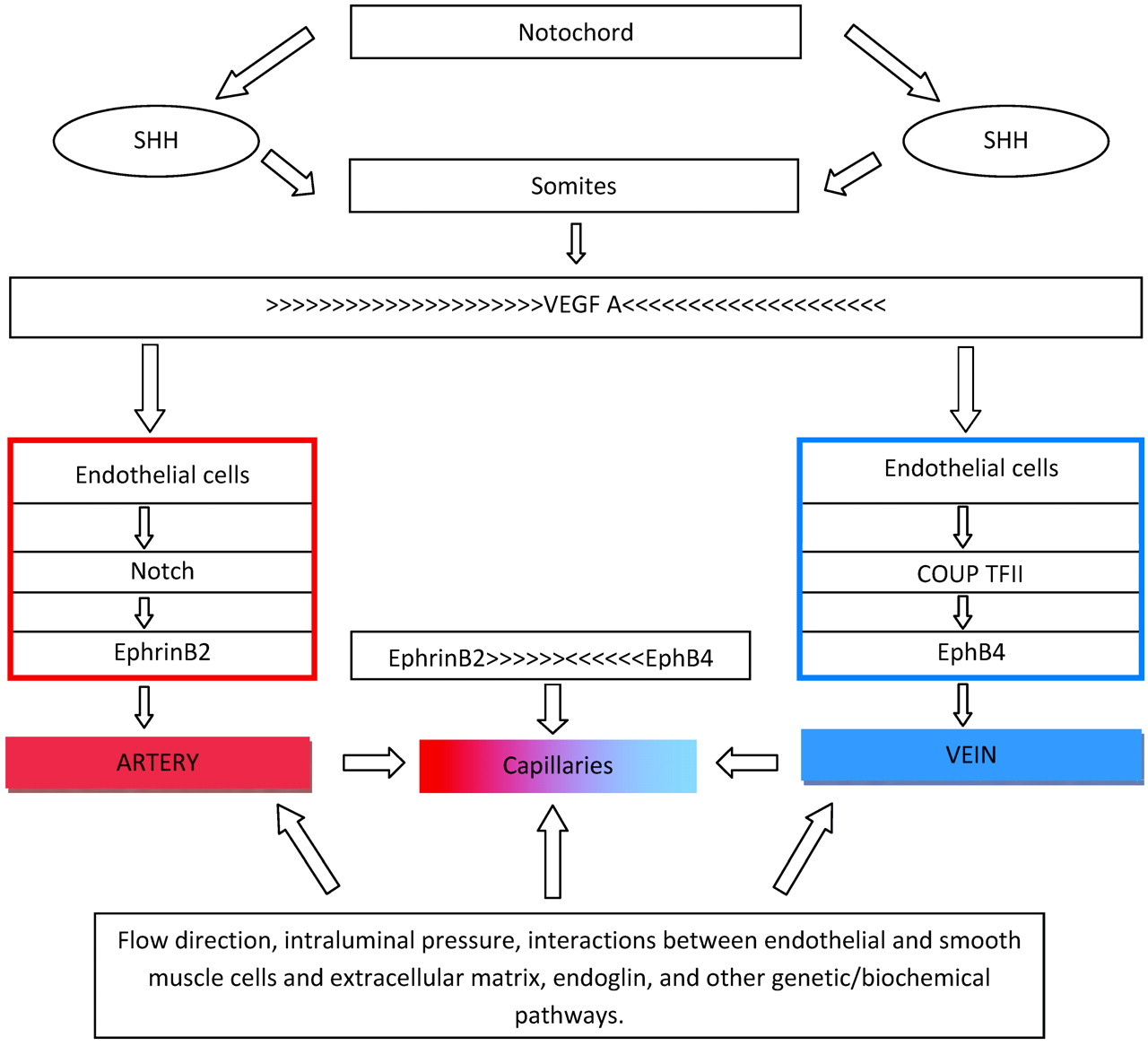
The process of vascular cell differentiation has been established through animal studies. Through a variety of mechanisms, sonic hedgehog induces tubulogenesis.3 Sonic hedgehog is expressed in the notochord and triggers endothelial cell formation but not cell-fate determination (Fig 1). When the function of hedgehog is affected, animals will not fuse their dorsal aortas; they lack differentiation of dorsal aortas from cardinal veins and have abnormal trunk circulatory systems that express only venous markers.1–4 If the function of sonic hedgehog is completely blocked, the aorta does not form. Sonic hedgehog induces expression of vascular endothelial growth factor A (VEGF-A) in somites, which in turn upregulates notch5 signaling, leading to a specific expression of ephrin-B2 in arteries and EphB4 in veins.1,3 VEGF-A is also involved in cell differentiation, proliferation, migration, and survival.3,4 VEGF-A is critical for vasculogenesis and postnatally for angiogenesis. Animals lacking VEGF-A or its receptor (VEGFR-2) develop few or no angioblasts and die early.3 Abnormalities of VEGF-A decrease arterial ephrinB4 and upregulate venous Flt4 (fms-related tyrosine kinase 4), leading to abnormal aortas.3 VEGF-A is highly expressed in peripheral nerves, and if deficient, nerves and accompanying arteries are abnormal or absent.5 It is thought that once an artery is formed, its smooth muscle cells direct the growth of adjacent sympathetic nerves. Once sympathetic neurons are established, they send axons to organs.5 Normal blood vessel differentiation occurs only if nerves are well aligned with arteries. Similarly, there is an isomer of VEGF that is involved in vein and lymphatic formation. Lymphatics originate from embryonic veins. Adult lymphatic endothelial cells are different from veins and arteries because they are not covered by a continuous basal membrane and their junctions are loose, allowing exchanges of interstitial and tissue fluids. Acquisition of lymphatic phenotype is regulated by VEGF, Prox1, cytoplasmic tyrosina kinase SYK, and SLP-76.6 Thus, we speculate that when this process is affected, it may lead to defects occurring simultaneously in both systems, such as venolymphatic malformations.

Sonic hedgehog (SHH) secreted by the notochord stimulates the somites to produce VEGF-A. Before cells form arteries or veins, their fate has been determined by a VEGF gradient and the presence of ephrinB2 (arteries) or EphB4 (veins). Balanced gradients of ephrinB2 and EphB4 result in capillary formation and maintenance. Once the circulatory system is established, a complex play between hemodynamics, oxygen concentration, intercellular communications, and genetic and biochemical actions aids in preserving blood vessel identity.
Let's summarize what we have learned up to this point: Arterial and venous destiny is determined by a molecular pathway involving sonic hedgehog, which subsequently affects notch and later VEGF-A (Fig 1). Loss of all, 2, or even 1 of these mechanisms results in lack of arterial identity. Because these 3 mechanisms are needed for arteries to develop, the default vessel identity was initially thought to be venous. However, high levels of COUP-TFII (chicken ovalbumin upstream promoter transcription factor II) and EphB4 are found in primitive veins but not in arteries; therefore, venous identity is probably the result of a dynamic cascade of events.4 Aberrations in these mechanisms result in abnormally large veins, malformed venous sinuses and cardinal veins, fusion of veins and arteries, and hemorrhage and edema leading to death.
Arterial progenitor cells express ephrinB2, whereas veins express EphB4 and other markers such as Flt4. These genes are part of the larger group called “Eph,” whose receptors modulate morphogenesis of various cell groups involved in the formation of the central nervous system. Eph also regulates cell migration and axonal guidance. Neuropilins are a form of VEGF, which is part of the gene family that controls axonal guidance in the brain.3 Both arteries and veins express neuropilin. Thus, one can start to understand that there is a relationship between vascular lesions (arterial and venous) and neuronal-migration abnormalities. Neuropilins are only 1 of many substances needed for axonal guidance. These substances work on the basis of attraction/repulsion and guide axons to final destinations. Once activation of Eph-related proteins has taken place, a variety of mechanisms that lead to cell changes begin. The EphB4 receptors need to be balanced with ephrinB2 ligands in order for vessels to acquire distinct identities and for postnatal morphogenesis to continue. If ephrinB2, expressed in endothelial cells, is knocked out, normal intercalations between arteries and veins do not happen and defective artery-to-vein connections arise. Thus, a balanced development of arteries and veins occurs only if gradient-like expression of ephrinB2 and EphB4 is present in endothelial cells. The interaction between ephrinB2 and EphB4 leads to formation of a hierarchically organized system of vessels that are more artery than vein in some regions, and, in others, more vein than artery. These connections vary in size and establish capillary networks. Maintenance of the arteriovenous interfaces also depends on ephrinB2 and EphB4. It is interesting to speculate that an imbalance between these substances may be the cause of various cerebral vascular malformations.
Notch receptors regulating early cell destiny are also involved in proliferation, apoptosis, maturation, and cell homeostasis. In humans, mutations in NOTCH3 or the NOTCH ligand (JAGGED-1) result in the vascular fragility seen with cerebral autosomal dominant arteriopathy (with subcortical infarcts and leukoencephalopathy) and Alagille syndrome, in which multiple arterial narrowings develop, respectively.4 Mice lacking notch genes exhibit life-compromising vascular anomalies. There are many other mechanisms that disrupt the function of notch genes, resulting in arterial deficiencies. COUP-TFII is a type of nuclear receptor that inhibits notch activation. By doing this, COUP-TFII suppresses receptors for VEGF-A. This suppression in turn causes abnormalities in the concentration of EphB4, which result in conversion of arterial-destined cell lineages to veins. Overexpression of an activated NOTCH receptor called int3 induces arteriovenous malformations (AVMs) postnatally in mice.1 By repressing the NOTCH receptors, arteries revert to normal, implying that treatment of AVMs may be possible this way.1
Vessel formation is complete when endothelial cells are surrounded by pericytes (in capillaries) or smooth muscle cells, which are present to varying degrees in larger arteries and veins. These processes are influenced by transforming growth factor β complex, which encompasses endoglin and ALK1 (activin receptorlike kinase 1). Endoglin is a glycoprotein found in endothelial and smooth muscle cells, which is critical for their development, morphology, and migration.7 Mutations of endoglin and ALK1 result in the loss of capillary beds and arteriovenous shunting and are thought to be the main abnormality in hereditary hemorrhagic telangiectasia types 1 and 2, respectively, syndromes characterized by AVMs in different organs and systems.
Once blood vessel identity is established, it needs to be perpetuated. Intraluminal hemodynamics and oxygen levels aid in maintaining vessel identity. However, arterial and venous identities are potentially reversible. Transplanted veins lose EphB4 and develop intimal-medial thickening to become more arterylike. This plasticity is predominantly seen in younger endothelial cells but may be induced in older ones if younger ones are grafted onto them. This aspect of identity maintenance is of importance for vessel transplantation.1–3
It is interesting to note that neo- and revascularization in postnatal life share many, if not all, of the above-described mechanisms and altering these is the basis of novel angiogenic or antiangiogenic therapies and, in the future, vessel-remodeling treatments. Blood vessels do not seem to have the luxury of asking themselves: to be or not to be? Their destiny is determined even before they are formed.
References
- Copyright © American Society of Neuroradiology
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.