
Abstract
BACKGROUND AND PURPOSE: In a large number of patients with Leigh syndrome (LS) and cytochrome c oxidase (COX) deficiency, mutations of the SURF-1 gene were recently identified. The aim of the present study was to review the MR findings in patients with LS to verify if the genetically homogeneous patients with LS and SURF-1 mutations (LS SURF-1 patients) had a homogeneous MR pattern that could be used to differentiate them from other patients with LS (LS non-SURF-1 patients).
METHODS: T1-, proton density-, and T2-weighted MR images of eight LS SURF-1 patients and 14 LS non-SURF-1 patients were reviewed. Enzymatic activity was determined according to standard methods. Genetic analysis was mostly performed by using stored DNA samples.
RESULTS: All LS SURF-1 patients had lesions in the brain stem and subthalamic nuclei. Six had lesions in the cerebellum. Only two had basal ganglial abnormalities. Ten LS non-SURF-1 patients had lesions in the brain stem, but in six they were mild. Ten patients had basal ganglial abnormalities (nine of 10 in the putamina). LS-SURF-1 patients had a more severe clinical course.
CONCLUSION: The MR pattern in LS SURF-1 patients is characteristic. Brain stem and subthalamic nuclei lesions may suggest the specific diagnosis. These patients die soon, probably because of lower brain stem involvement. Basal ganglial abnormalities are common only in LS non-SURF-1 patients. The absence of putaminal lesions, therefore, does not exclude the diagnosis of LS.
Leigh syndrome (LS), or subacute necrotizing encephalomyelopathy, is an inherited, progressive, metabolic disease of infancy and childhood. As first described by Denis Leigh in 1951 (1), it causes striking neuropathologic features of focal, bilateral, and symmetric necrotic lesions associated with demyelination, vascular proliferation, and gliosis in the brain stem, diencephalon, basal ganglia, cerebellum, and (occasionally) cerebral white matter. Clinical signs usually appear within the patient’s first few years of life and consist of hypotonia, failure to thrive, psychomotor regression, and brain stem and basal ganglia dysfunction that results in ataxia, ocular movement abnormalities, dystonia, and swallowing and respiratory disturbances. The age at onset and clinical features may vary widely among patients; in most, the disease has a progressive course and eventually leads to death. Pathologic similarity with other metabolic encephalopathies (ie, Wernicke syndrome) and the common finding of increased lactate levels in the plasma and CSF suggested a metabolic nature of the disease, as later confirmed by the finding of a severe deficiency in mitochondrial adenosine triphosphate (ATP) production. Although initial observations suggested an autosomal recessive inheritance, later reports of autosomal dominant, X-linked, and maternally inherited cases indicated that causative genes exist in both nuclear and mitochondrial genomes. Genetic heterogeneity has been confirmed with the identification of functional or molecular defects in several enzyme systems involved in mitochondrial energy production, including the pyruvate dehydrogenase complex (PDHC), respiratory chain complexes I–IV (cytochrome c oxidase [COX]), and mitochondria-encoded ATPase 6 subunit of complex V (2). Such striking clinical, molecular, and biochemical variability accounts for the preferential use of the term Leigh syndrome instead of Leigh disease.
Diagnosis in vivo is accepted when a combination of appropriate clinical features, lactic acidosis, and congruent abnormalities on CT and MR studies are present (3).
A number of investigators (3–7) have discussed the imaging findings in LS and have tried to correlate a specific enzymatic defect to a homogeneous radiologic pattern, with no success.
In a large number of LS patients with a profound, isolated, and generalized COX defect, loss-of-function mutations in SURF-1, a gene located on chromosome arm 9q34, have been recently identified (8, 9). The protein product of SURF-1 plays an important although largely unknown role in the assembly of complex IV.
Before the discovery of SURF-1 mutations, the frequent association of lesions of the subthalamic nuclei with COX deficiency was reported (5, 6).
To verify that SURF-1 mutations correspond to a homogeneous MR pattern, in other words, that the genotype and its phenotypic radiologic expression are correlated, we reviewed the MR studies of 22 patients with LS: eight with LS with COX deficiency and SURF-1 mutations (LS SURF-1 patients) and 14 with LS due to other genetic conditions (LS non-SURF-1 patients). The demonstration of such a correlation might shorten the diagnostic process.
Methods
The 22 selected children (nine male, 13 female) are part of a larger series of 330 cases with a positive definite diagnosis of mitochondrial disease that has an onset in infancy and adulthood. All 22 cases were of Italian origin, and all patients presented in their first 2 years of life with psychomotor regression and signs of brain stem and basal nuclei involvement associated with lactic acidosis and congruent signal intensity abnormalities on MR images. All children were examined by means of serial neurologic examinations, electrophysiological studies (including electroencephalography [EEG], peripheral nerve conduction study, multimodal evoked potential study, and electroretinography), ophthalmologic examinations, and metabolic evaluations (including determinations of plasma and CSF lactate and pyruvate levels, blood gas analysis, analysis of urinary organic acids). Patients were tested by using standard psychometric scales (Griffiths, Wechsler Preschool and Primary Scale of Intelligence [WPPSI], Wechsler IntelligenceScale for Children-Revised [WISC-R]). On the basis of the degree of lactic acidosis, the patients were given sodium bicarbonate to correct metabolic decompensation. Three children with complex I deficiency were also treated with sodium dichloroacetate, which reduces lactic acidosis in mitochondrial disorders (10), particularly in complex I deficiency.
Morphologic examination of skeletal muscle tissue included histologic analysis with modified Gomori trichrome staining for ragged red fibers, ATPase staining to assess myofibrillar integrity and muscle-type fiber predominance and distribution, and different histochemical stains for oxidative enzymes (COX and succinate dehydrogenase [SDH]). Biochemical assays of respiratory chain complexes activity were performed on muscle homogenate and digitonin-treated fibroblasts by using spectrophotometric methods (11). Specific activity of each complex was then normalized for citrate synthase, an indicator of mitochondrial number. All subjects with COX deficiency were screened for mutations in the SURF-1 gene by means of genomic DNA polymerase chain reaction (PCR) amplification and sequencing (8).
MR studies were performed from 1988 to 2000, with different units operating at 0.5 and 1.5 T, sometimes at different institutions. All the studies included T1-, proton density-, and T2-weighted spin-echo images that, at least in one plane, covered the whole brain. Images obtained with other sequences were available in some patients and included gradient-echo, inversion-recovery (IR), fluid-attenuated inversion recovery (FLAIR), and turbo spin-echo (TSE) images. Contrast material-enhanced studies were obtained in one LS non-SURF-1 patient (case 13) with a tumorlike swelling of the midbrain, as depicted on the initial MR image.
The eight patients with LS-SURF-1 (four male, four female) were aged 1–4 years when the first MR study was performed. Only two patients underwent repeated MR imaging: one after 4 months (patient 2) and the other after 6 years (patient 7).
The 14 patients in the LS non-SURF-1 group included four patients with COX deficiency without SURF-1 mutations, six with defects of other respiratory chain complexes (complex I in four, complex III in one, and multiple complexes in one), two patients with PDHC deficiency, one with a T8993G mutation in mitochondrial DNA (neurogenic muscle weakness, ataxia, and retinitis pigmentosa [NARP] and maternally inherited LS [MILS]), and one with an undefined biochemical defect. Five were male and nine were female, ranging in age from 7 months to 9 years at the time of the first MR examination. Seven patients underwent repeated MR studies (range, 1–8 years from the first examination).
Results
Clinical Findings
In the LS-SURF-1 group, clinical features were homogeneous. The age at onset was 9–16 months. Vomiting and failure to thrive were the first symptoms, and neurologic deterioration occurred rapidly, with severe hypotonia, ataxia, abnormalities of eye movements, and early respiratory impairment. Peripheral neuropathy was invariably present, whereas seizures and extrapyramidal signs were seldom observed. Compared with the severity of motor regression, cognitive functions were well preserved. None of the patients had microcephaly. Patient 7 had the same spectrum of symptoms but had a milder clinical course and was still alive at this writing. Patient 2 was surviving with permanent respiratory assistance. All the other patients of this group died from respiratory failure before they were aged 5 years.
More heterogeneous clinical features were observed in the LS non-SURF-1 patients. The age at the onset of symptoms ranged from birth to 24 months of age. Moderate-to-severe mental impairment was always present, whereas spasticity and dystonia characterized the motor disability more than ataxia and hypotonia. Ten of the 14 patients had poor growth, 10 had abnormal eye movements, six had seizures requiring antiepileptic drugs, four had microcephaly, and one had profound deafness. Three of the LS non-SURF-1 patients died by age 2 years, whereas the other 11 patients were still alive.
MR Findings
The lesions described were those seen on initial MR images. The appearance or disappearance of lesions on follow-up MR images is indicated in the text and Tables 1 and 2. The lesions were clearly visible and hyperintense on proton density- and T2-weighted images; they were hypointense, and usually less evident, on T1-weighted images.
LS SURF-1 Group
LS Non-SURF-1 Group
LS-SURF-1.—All eight patients had lesions in the brain stem: eight in the medulla oblongata, seven in the pontine tegmentum, and eight in the midbrain. In the medulla, the lesions were moderately or markedly extensive in five patients; the lesions were generally centered on the inferior olives and sometimes involved the area of the nucleus of the solitary tract (Fig 1A). The pons was involved only in the tegmentum, with symmetric abnormalities that were often centered on the central tegmental tract and the reticular formation (Fig 1B). In the midbrain, the periaqueductal area was always involved (Fig 1C); the substantia nigra was affected in three patients; the red nucleus, in two; and the tectum at the level of the inferior colliculi in two.

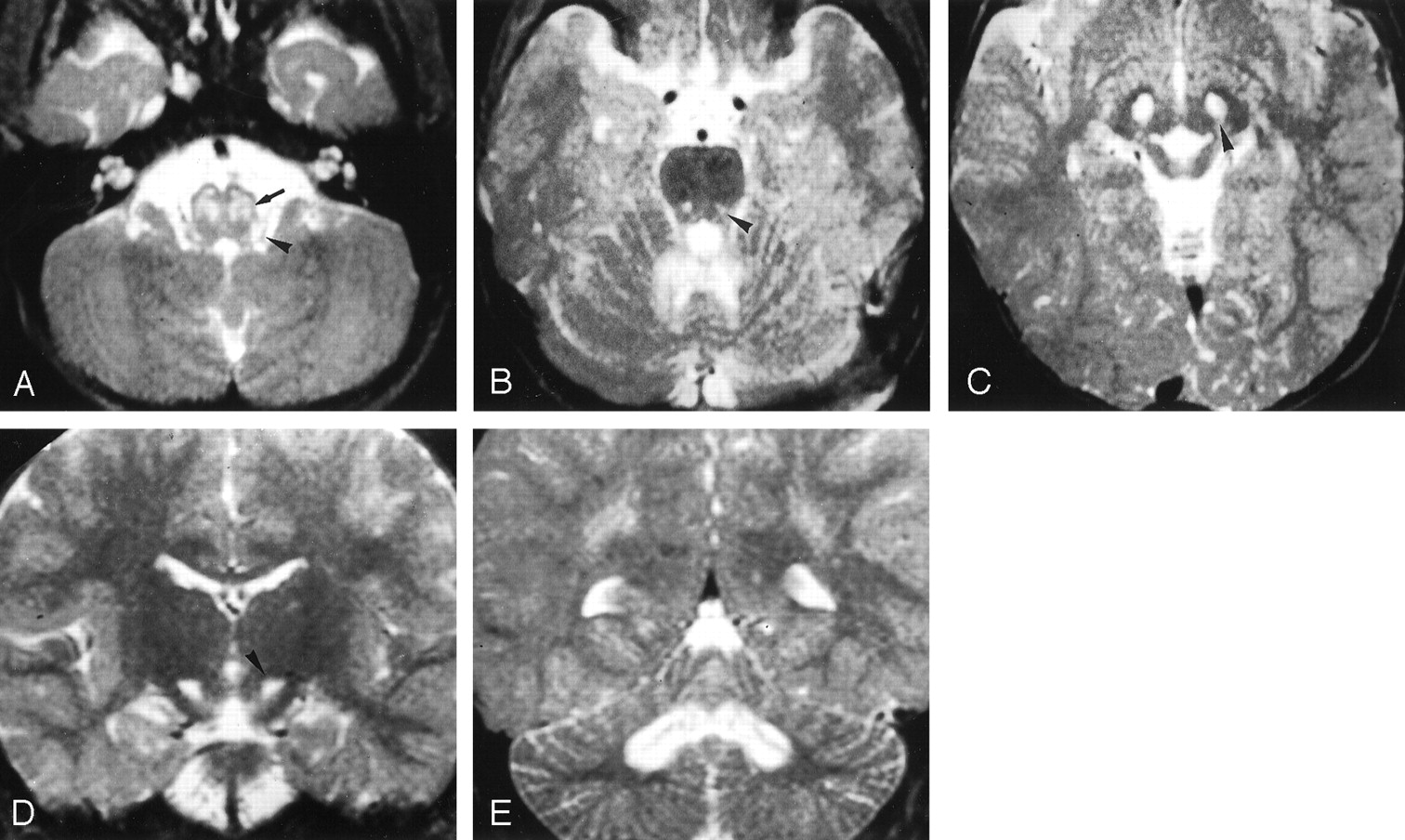
LS SURF-1. Case 5. Male patient, 2 years old.
A–C, Axial T2-weighted images (3000/120/1 [TR/TE/NEX]) show hyperintense lesions involving the region of the inferior olivary nuclei (arrow in A) and the dorsolateral medulla at the base of the restiform bodies (arrowhead in A), punctuate lesions in the pontine tegmentum (arrowhead in B) and more extensive abnormalities in the cerebellar white matter, and lesions in the periaqueductal area and subthalamic nuclei (arrowhead in C).
D and E, Coronal T2-weighted sections (3000/120/1) confirm the presence of lesions in the subthalamic nuclei (arrowhead in D) and show extensive white matter involvement of the cerebellum centered on the dentate nuclei (in E).
Involvement of the cerebellum was marked in six patients, doubtful in two, and always centered on the dentate nuclei with extension to the surrounding white matter in five (Fig 1E). In all eight patients, the subthalamic nuclei were abnormal (Fig 1C and D). Only two patients had lesions of the lenticular nuclei that involved the posterior part of the putamen. Two patients had small symmetric lesions in the medial part of the thalami. In one patient (case 6), high signal intensity was observed on T2-weighted images in the parietal white matter. In patient 2, follow-up MR demonstrated mild worsening of pontine and medullary lesions, which was correlated to a clinical worsening. In patient 7, who was the oldest patient of this group and who had the mildest clinical course, follow-up MR images obtained after 6 years showed regression of the lesions in the medulla, cerebellum, and thalami. Images also showed the disappearance of lesions in the subthalamic nuclei, whereas the putaminal abnormalities mildly progressed. The distribution of the lesions is summarized in Table 1.
LS Non-SURF-1.—Ten patients had lesions in the brain stem, which were barely visible and had modest extension, except for those in four patients (cases 1, 8, 10, 13) who presented with an extensive midbrain-diencephalic involvement (Fig 2A and B). In one of these patients (case 13), midbrain involvement resulted in an unusual pseudotumoral appearance with asymmetric, marked swelling of the midbrain and focal nodular enhancement. Four patients had lesions in the cerebellum, in the dentate nuclei, surrounding white matter, or both. One of them (case 10) also had cortical cerebellar abnormalities. Five patients had lesions in the central-medial or posterior-medial part of the thalami, four of them bilateral (Fig 2B), and one unilateral; in one patient (case 10), diffuse supratentorial lesions were present with more extensive thalamic abnormalities. Ten patients had lesions in the basal ganglia (nine lesions in the putamen, five in the caudate nuclei, three in the pallida). Two patients (cases 2 and 10) had extensive abnormalities in the white matter of the cerebral hemispheres; in one (case 10), a multicystic appearance was present throughout the brain.

LS non SURF-1. Case 8. Female patient, 8 months old at first examination.
A and B, Axial T2-weighted MR images (3000/120/1) obtained at first examination show hyperintense lesions in the substantia nigra (arrow in A) and medial thalamic nuclei (arrowhead in B). The globi pallidi and white matter, still unmyelinated, are slightly hyperintense, with a tiny, focal hyperintensity in the left pallidum.
C, Follow-up MR image (2028/120/2), obtained 2 years and 2 months later, shows lesions in the basal ganglia that involve both the putamina and the head of the left caudate nucleus. A minimal residual right thalamic lesions is visible (arrowhead). Moderate diffuse brain atrophy is present.
Seven patients had one or more follow-up MR imaging sessions. In one patient (case 7), the lesions were unchanged. In three patients (cases 3, 4, 14), the signal intensity abnormalities in the basal ganglia became more sharply defined, and the basal ganglia shrank. In one (case 9), a new lesion in the head of the caudate nuclei appeared, and lesions in the putamina became more extensive. In two patients treated with dichloroacetate (cases 8 and 13), the distribution of the signal intensity abnormalities changed, with the nearly complete disappearance of the extensive mesencephalodiencephalic lesions within a few months, the appearance of transient abnormalities in the subthalamic nuclei, and the late development of lesions in the basal ganglia (after 15 months in case 8 and after 4 years in case 13) (Fig 2C). In the third case treated with dichloroacetate (case 1), follow-up MR images obtained at another hospital were reported to show reduced basal ganglial abnormalities. The distribution of the lesions is summarized in Table 2.
Discussion
Involvement of the basal ganglia, principally the putamen, has long been considered the hallmark of LS (4, 7) particularly with CT studies, although previous neuropathologic reports emphasized that brain stem and diencephalic abnormalities are a nearly constant and characteristic features (12). With MR imaging, recognition of involvement of the brain stem and cerebellum has become more common (3, 5, 13). The symmetric involvement of the subthalamic nuclei was common although not invariably present in LS patients with COX deficiency (5, 6).
In our LS SURF-1 patients, the MR pattern is uniform, with constant involvement of the brain stem and subthalamic nuclei and nearly constant involvement of the cerebellum, whereas putaminal involvement was rare (two [25%] of eight cases) (Table 1).
In LS non-SURF-1 patients, involvement of the basal ganglia was the most common or prominent finding (12 [83%] of 14 cases). Brain stem lesions, although they were found in 10 (71%) of the 14 cases, were usually mild. Subthalamic involvement was rare and transient (two [14%] of 14 cases). In the four patients of our series with extensive involvement of the brain stem, the bulk of the lesions was located in the midbrain and extended cranially to the diencephalon. In two of these patients, follow-up MR images showed nearly complete regression of the lesions and showed the late appearance of basal ganglia abnormalities (Table 2).
We had control MR images in only two (25%) LS SURF-1 patients, but we had follow-up MR images, often multiple studies, in seven (50%) of the 14 LS non-SURF-1 patients. This difference may simply be due to the fact that LS SURF-1 patients die sooner. The more severe clinical course with the increased incidence of respiratory failure and death in LS SURF-1 patients may be explained by the constant involvement of the brain stem in this group. In particular, severe respiratory failure and blood pressure imbalances have been associated with involvement of the lower brain stem (13) and the region of the nucleus of the solitary tract (14). All the LS SURF-1 patients had lesions in the medulla oblongata, whereas only five of the 14 LS non-SURF-1 patients had tiny lesions in this segment of the brain stem. In patient 7, the only long-term survivor among our LS SURF-1 patients, exhibited the brain stem lesions slightly regressed.
Identifying LS SURF-1 patients is relevant for prognosis, genetic counselling, and prenatal diagnosis. Recognition of the characteristic MR imaging patterns described here shortens the diagnostic process in affected patients.
The distribution of the lesions in the brain stem and cerebellum described here is not unique to LS, because it appears similar to the distribution of abnormalities observed in an outbreak of enteroviral encephalomyelitis caused by enterovirus 71 (15). Some indications suggest that, in certain conditions, even toxic agents may damage the nervous system by interfering with the mitochondrial function; therefore, attention to serendipitous findings (eg, presence of lactate in the white matter of patients with leukoencephalopathy caused by heroin inhalation [16]) may offer a clue for new and interesting pathogenetic insights. These speculations are also supported by the neuropathologic and radiologic observations of brain stem lesions in the region of the solitary tract in heroin sniffers (17); this area was involved in a few of our LS SURF-1 patients.
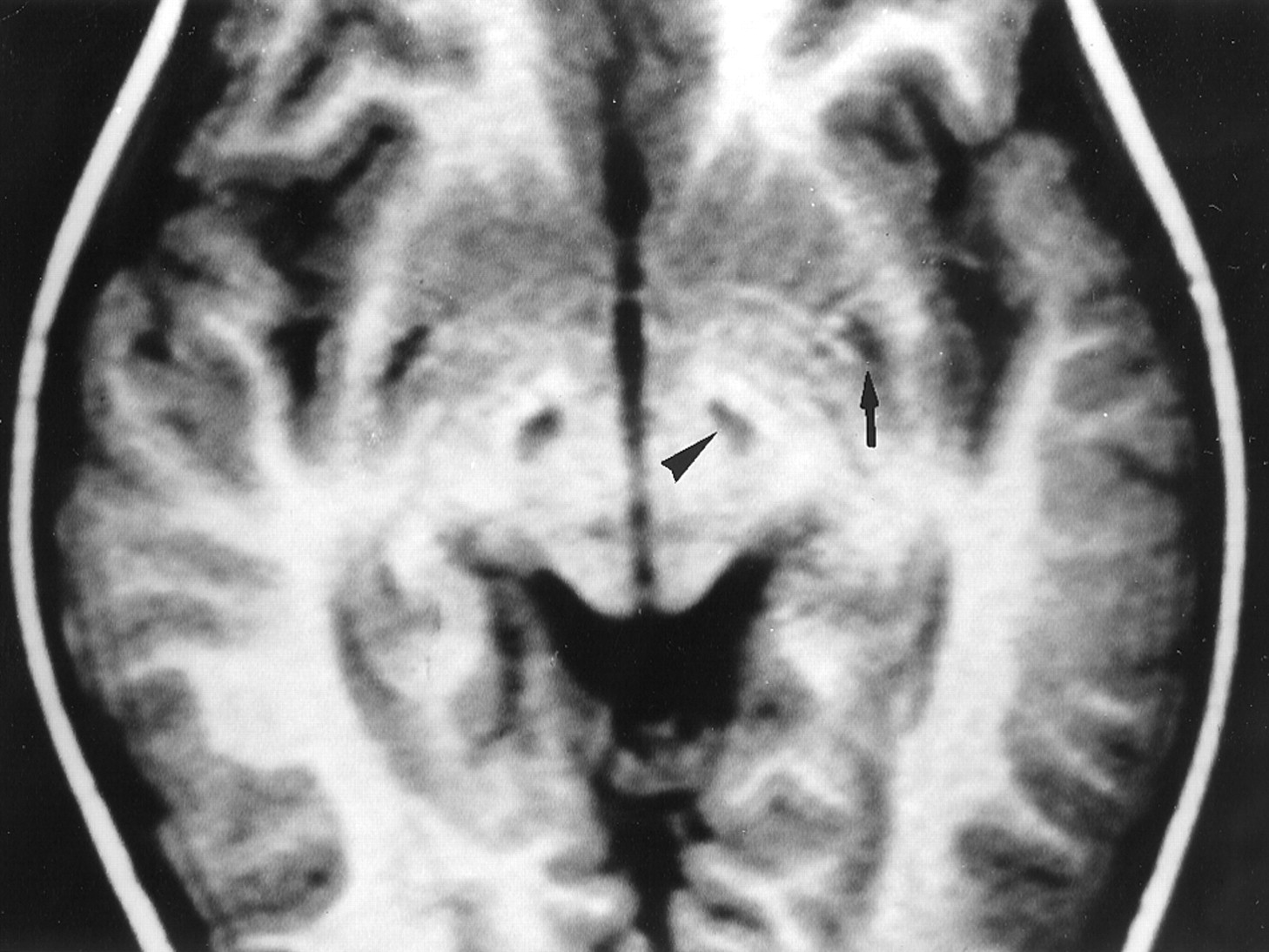
In our experience, LS SURF-1 is the only disorder with constant bilateral involvement of the subthalamic nuclei. Unilateral lesions involving the subthalamic nuclei are seen in several disorders (vascular, infectious, etc), usually associated with hemiballismus (18). In these cases, a larger lesion affects the nucleus. In our cases, the lesions were bilateral and neatly confined to the subthalamic nuclei (Fig 3). None of our patients presented with ballismus, possibly because of the slow development of the lesions or because of the involvement of other structures, such as the red nucleus or pyramidal tract, which must be preserved to cause development of ballismus (6).

LS-SURF-1. Case 7. Male patient, 4 years old.
Axial IR T1-weighted MR image (1582/80/2; inversion time, 750 ms) shows sharply defined hypointense lesions of the subthalamic nuclei (arrowhead) and hypointense posterior putaminal lesions (arrow). On follow-up MR images obtained 6 years later (not shown), the former disappeared, and the latter progressed.
We observed bilateral subthalamic lesions in only one other patient with a mitochondrial encephalopathy similar to Kearns-Sayre syndrome but never in patients with other metabolic diseases.
Unlike our series, recent studies (19, 20) of seven LS-SURF-1 patients revealed extensive abnormalities in the basal ganglia and, occasionally, the midbrain and dentate nuclei abnormalities, but no detailed MR findings are described. In another case, only brain stem and cerebellar lesions are mentioned (21). A leukodystrophic aspect, particularly in the posterior cerebral regions with cyst formation, was reported in a case (22) with a novel mutation in the SURF-1 gene; this patient, however, also had lesions in the cerebellum and lower brain stem, but no lesions in the basal ganglia. Therefore, the spectrum of the MR abnormalities is broader than it appears in our series, even in a genetically determined group of patients such as LS SURF-1 patients.
Conclusion
In our opinion, to date, the MR demonstration of symmetric brain stem and subthalamic nuclei involvement indicates a mitochondrial disorder, and when COX deficiency is demonstrated, it should prompt the search for SURF-1 genetic mutations. Moreover, we have demonstrated that the involvement of basal ganglia is an inconsistent finding in LS; in fact, it may be absent in a substantial number of patients. Therefore, we suggest that the absence of putaminal lesions should no longer be considered an exclusion criterion for the neuroradiologic diagnosis of LS.
Footnotes
Partly supported by Telethon grant no. 1180 and a grant from the Fondazione Pierfranco e Luisa Mariani.
Presented in part at the 10th meeting of the European Neurological Society, Jerusalem, Israel, June 18–22, 2000, and at the XVII National Congress of the Italian Association of Neuroradiology, Genova, Italy, December 14–17, 2000.
References
- Received July 30, 2001.
- Accepted after revision March 5, 2001.
- Copyright © American Society of Neuroradiology
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Leigh Syndrome in Drosophila melanogaster: MORPHOLOGICAL AND BIOCHEMICAL CHARACTERIZATION OF Surf1 POST-TRANSCRIPTIONAL SILENCING
- Bilateral hypertrophic olivary nucleus degeneration on magnetic resonance imaging in children with Leigh and Leigh-like syndrome
- SURF1 deficiency causes demyelinating Charcot-Marie-Tooth disease
- A common pattern of brain MRI imaging in mitochondrial diseases with complex I deficiency
- Post-transcriptional Silencing and Functional Characterization of the Drosophila melanogaster Homolog of Human Surf1
- Functional and genetic studies demonstrate that mutation in the COX15 gene can cause Leigh syndrome
- MR Imaging in Human Rabies